Growth plate extracellular matrix defects and short stature in children
Article information

Abstract
Many etiological factors causing short stature have already been identified in humans. In the last few years, the advent of new techniques for the detection of chromosomal and molecular abnormalities has made it possible to better identify patients with genetic causes of growth failure. Some of these factors directly affect the development and growth of the skeleton, since they damage the epiphyseal growth plate, where linear growth occurs, influencing chondrogenesis. In particular, defects in genes involved in the organization and function of the growth plate are responsible for several well-known conditions with short stature. These genes play a pivotal role in various mechanisms involving the extracellular matrix, intracellular signaling, paracrine signaling, endocrine signaling, and epigenetic regulation. In this review, we will discuss the genes involved in extracellular matrix disorders. The identification of genetic defects in linear growth failure is important for clinicians and researchers in order to improve the care of children affected by growth disorders.
Highlights
· Chondrodysplasias are rare genetic diseases caused by various etiologies involving more than 450 genes, characterized by severe harmonic or disharmonious short stature, skeletal deformities, craniofacial anomalies, and premature joint degeneration.
· These genes encode for extracellular proteins, such as collagen, proteoglycans, hyaluronan, and many other specific proteins that represent the structural components of the extracellular matrix, which has a critical role in the structural support of chondrocytes and represents a medium for signaling molecules and growth factors.
· New molecular genetics techniques can recognize an increasing number of new genes implicated in short stature, allowing clinicians and researchers to improve the care of children affected by growth disorders.
Introduction
Human height or stature is a somatic trait that has a normal distribution, with a small fraction (3%–5%) of individuals exhibiting extreme short or tall height phenotypes. Short stature is defined as height that is at least 2 standard deviations (SD) below the mean of a specific population adjusted for age, sex, and pubertal stage [1]. The severity of growth failure indicates the likelihood of pathology, which is very low (below 2%) with a height standard deviation score (SDS) above -2 (around the third percentile) and increases to around 50% between -2 and -3 and to 80% beyond -3 SDS (0.1 percentile) [2]. According to the International Classification of Pediatric Endocrine Diagnosis, short stature may be idiopathic (in which no possible cause is identified), secondary to organ system disease (e.g., chronic kidney disease) or to environmental factors, or may arise from genetic mutations (primary short stature) [3].
Cartilage extracellular matrix
A wide range of factors may cause a child's reduced longitudinal growth, resulting in short stature in adulthood. Some of these factors influence chondrogenesis in the epiphyseal growth plate and affect the development of the skeleton, which is the main determinant of height [4]. The growth plate is the cartilaginous structure between the epiphysis and metaphysis where linear growth occurs [5].
In this region, there are 3 cell populations organized into 3 different zones with peculiar characteristics [6]. The resting zone is made up of round chondrocytes known as prehypertrophic chondrocytes. The proliferating zone is divided into the prehypertrophic zone, containing the maturing chondrocytes organized in longitudinal columns, and the hypertrophic zone, where the cartilage gradually goes through ossification (Fig. 1) [7].
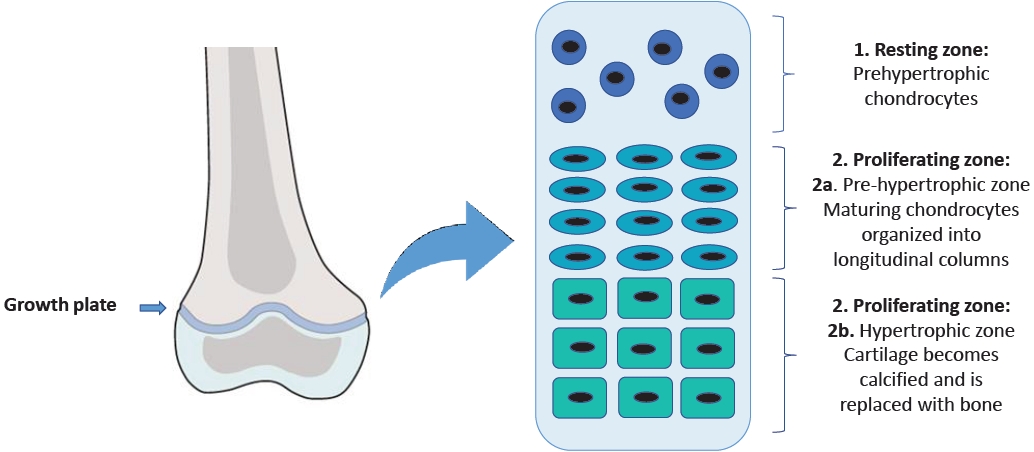
The structure of the growth plate.
While chondrocytes undergo this process of proliferation and differentiation, their endoplasmic reticulum (ER) synthesizes extracellular proteins, such as collagen, proteoglycans, hyaluronan, and many other specific proteins that represent the structural components of the extracellular matrix (ECM) [8,9]. ECM has a critical role in the structural support of chondrocytes and represents a medium in which signaling molecules and growth factors are able to spread through the avascular cartilage toward the target cells [8,10]. Therefore, it is obvious that mutations in genes that regulate the ECM may cause isolated short stature or severe skeletal dysplasia. Many genetic causes of growth disorders have already been established, but recent genomewide studies have highlighted new monogenic causes of growth disorders [4].
One mechanism that may lead to ECM alterations is the so-called "endoplasmic reticulum stress," which determines the accumulation of folded proteins [11] and can cause altered proliferation and differentiation of the chondrocytes of the growth plate with consequent skeletal diseases, such as chondrodysplasias [12].
Chondrodysplasias represent a heterogeneous group of rare genetic diseases caused by various etiologies involving more than 450 genes [13]; as a group, the incidence of chondrodysplasias is 1 per 5,000 births [14]. They are multisystem disorders whose main characteristic is represented by severe short stature, often associated with skeletal deformities, craniofacial anomalies, and premature joint degeneration. Additionally, these disorders may be associated with involvement of other organs, including neurological, auditory, visual, pulmonary, cardiac, renal, and psychological complications [14].
In this review, we aim to describe the main causes of short stature, focusing on the genes involved in ECM disorders (Table 1).

Extracellular matrix genetic disorders involved in short stature
Collagens (COL2A1, COL10A1, COL9A2, COL11A1, COL27A)
Collagens represent one of the primary proteins in the ECM. Mutations in collagen genes are involved in many diseases like chondrodysplasias [15].
COL2A1 gene (12q13.1-q13.2) encodes type II collagen, a fibrillar protein of 1,487 amino acids. Type II collagen is the main component of hyaline cartilage ECM. In the growth plate, it is synthesized by the chondrocytes of the proliferation zone [16]. Type II collagen is composed of 3 identical α1-polypeptide chains made up of 1060 amino acids. Type II collagen molecules self-assemble into cross-linked fibrils which form the type II collagen fibers. These fibers interact with other macromolecules, forming the ECM of cartilage [17].
Mutations in the COL2A1 gene cause a wide variety of rare autosomal dominant diseases. More than 400 mutations have been described. These mutations are associated with an accumulation of immature procollagen in the cisternae of chondrocytes rough ER, causing cellular stress [18]. Furthermore, mutated type II collagen molecules would not be able to selfassemble into fibrils, which are necessary to allow for the correct columnar arrangement of the chondrocytes in the growth plate or to interact correctly with the other macromolecules of the matrix [19].
Mutations of this gene are responsible for spondyloepimetaphyseal dysplasia (SEMD) and spondyloepiphyseal dysplasia (SED) congenita, which are characterized by delayed ossification of the vertebrae and pubic bones with subsequent dwarfism, kyphoscoliosis in early childhood, and shortening of long bones. In spondyloperipheral dysplasia, patients show an important short stature (mean length 45 cm at term) associated with the absence of ossification in pubic bones, distal femoral epiphyses, and cervical and sacral vertebras with consequent lumbar lordosis.
COL10A1 (6q22.1) encodes collagen X, a homotrimer collagenous protein formed by 3 α-1 (X) chains. Type X collagen is expressed only in the hypertrophic chondrocytes layer of the cartilage growth plate.
Schmid's metaphyseal chondrodysplasia (SMCD) is inherited in autosomal dominant manner or with a novo mutation phenotype and seems to be related to an abnormal assembly of collagen trimer in the ER of hypertrophic chondrocytes, due to mutations in COL10A1 C-terminal noncollagen domain (NC1), which determines ER stress and reduced levels of functional type X collagen [20].
SMCD is typically diagnosed in early childhood and is characterized by progressive short stature starting from 2 years (adult height is usually more than 3.5 SD below the mean) associated with genu varum and waddling gait. Facial features and head size are normal, and there are no extraskeletal manifestations [21].
COL9A2 encodes type IX collagen, a heterotrimer fibrillar protein of the ECM. Each peptide chain consists of 3 collagenous domains separated by 4 noncollagenous domains [22]. Several mutations causing a loss of amino acids in the third collagenous domain are involved in the pathogenesis of multiple epiphyseal dysplasia (MED) [23].
MED is a skeletal dysplasia which, genetically, can be related to mutations in COMP, MATN3, COL9A1, COL9A2, and COL9A3, characterized by different clinical features. Clinical progress associated with COL9-MED is relatively mild, showing joint disease but only sometimes altered growth. Epiphyseal ossification delay and alterations in their shape are the main radiographic signs [24]. The MED phenotype usually manifests after the first 1–2 years of life. One of the first signs is joint pain, especially after exercise, affecting the hip and knee joints. On the contrary, growth failure is slowly progressive and can lead to mild to moderate short stature (approximately below the third percentile) at the age of 5–6 years [25].
COL11A1 and COL27A1 play a decisive role in the organization of the growth plate proliferative zone. Mutations in these proteins are associated with several forms of skeletal dysplasia [26-28]. Heterozygous mutations in the COL11A1 gene cause Stickler's syndrome [29] and Marshall's syndrome, which are phenotypically similar to each other, involving ophthalmic, articular, orofacial, and auditory defects. Radiographically, joint surface irregularities have been described [30,31]. Homozygous mutations of COL11A1 cause fibrochondrogenesis 1, a skeletal dysplasia presenting with short limbs, flat midface, and protrudent abdomen [32].
The COL27A1 gene encodes for collagen type XXVII, proalpha 1, and is located on human chromosome 9q32-33. The protein consists of 1860 amino acids and plays a pivotal role in the organization of ECM [33].
Recessive mutations in the COL27A1 gene are responsible for a rare genetic disorder called Steel syndrome [34,35], which is characterized by mesomelic short stature, bilateral genua valga, absence of capital femoral epiphyses, shallow bilateral acetabula, incomplete ossification of pubic bones, scoliosis, pectus excavatum, and facial dysmorphism [36].
Fibrillins
Fibrillins (FBN1) are structural proteins that, like collagen, form fibers and interact with many other connective tissue molecules [37]. In the human genome, 3 different genes (FBN1, FBN2, and BN3) encode fibrillins; FBN1 and FBN2 are mainly involved in growth regulation [38]. The human FBN1 gene is located on chromosome 15q15 21.1 and comprises 65 exons. FBN1 encodes fibrillin-1, a calcium-binding protein composed of 2871 amino acids that forms 10–12 nm microfibrils in the ECM [39].
Fibrillin microfibrils play a pivotal role in the structural integrity of many organs, such as the aortic wall and the suspensory ligament of the lens [37]. FBN1 mutation causes several genetic connective tissue diseases called fibrillinopathies [40], such as Marfan syndrome, an autosomal dominant disorder characterized by tall stature, skeletal and ocular abnormalities, cardiovascular involvement [41], and acromelic dysplasias characterized by short stature, short extremities, and joint stiffness. Acromelic dysplasias include Weill-Marchesani syndrome (WMS), geleophysic dysplasia, acromicric dysplasia (AD), and Myhre syndrome. The first condition in which FBN1 mutation has been observed was WMS [42], which is caused by an in-frame deletion of 24 nucleotides in exon 41 [43], while missense mutations in exons 41 and 42 cause geleophysic dysplasia and AD [44]. In few cases, an in-frame deletion of exons 9–11 may also be responsible for WMS [45].
Currently, 4 types of WMS have been described: type I, caused by a homozygous mutation in the ADAMTS10 gene; type II, caused by a heterozygous mutation in FBN1; and types III and IV, which are due to homozygous mutations in LTBP2 and ADAMTS17, respectively [43].
The primary clinical features of WMS are proportionate short stature, brachydactyly, joint stiffness, ocular abnormalities, and cardiac involvement [46]. Although some of these alterations such as ophthalmic abnormalities have been extensively described, there is still no detailed information in the literature regarding short stature and a possible correlation with growth hormone (GH) deficiency or whether these patients respond to GH therapy [46]. Short stature is reported in all affected individuals and appears in the first years of life. The average height of an adult male with WMS is approximately 142–169 cm and that of an adult female is 130–157 cm [47].
Geleophysic dysplasia is a severe form of acromelic dysplasia. Two types of geleophysic dysplasia have been described: type I, due to an ADAMTSL2 recessive mutation [48], and type II, caused by autosomal dominant mutations in FBN1 [49]. The term "geleophysic" derives from geleos, which means happy, and physis, which refers to nature. In fact, patients present with a "happy" face, with full cheeks, small palpebral fissures, short nose with anteverted nares, long smooth philtrum, and thin vermilion border of the upper lip [50]. Geleophysic dysplasia is characterized by short stature (<3 SD), brachydactyly, dysplastic femoral epiphysis, hepatomegaly, joint stiffness, progressive cardiac valvular thickening, and tracheal stenosis with bronchopulmonary insufficiency [51].
The main characteristics of AD are severe short stature, joint stiffness, shortened hands and feet, and mild facial dimorphism without cognitive impairment. Patients have normal height at birth but progressively decrease in centiles through childhood [44].
Aggrecan
Aggrecan (ACAN), the most important proteoglycan of growth plate cartilage, is encoded by the ACAN gene, which is located in chromosome 15q26.1 [52]. It is composed of an N-terminal domain, 2 globular domains (G1 and G2), 2 interglobular domains (CS and KS), a selectin-like domain (G3), and a C-terminal domain [53,54].
Aggrecan-type SEMD is caused by an autosomal recessive mutation in the G3 domain of aggrecan (p.Asp2267Asn) [55] and represents a severe skeletal dysplasia characterized by extreme short stature (final stature of 66–71 cm). Other clinical features include dysmorphic craniofacial abnormalities, brachydactyly, barrel chest, and mild lumbar lordosis [56]. Radiographic examination showed irregular epiphyses and enlarged metaphyses, particularly in the knees.
SED, Kimberley type, is a less severe skeletal dysplasia caused by an autosomal dominant mutation, resulting in the creation of a premature stop codon (p.Gly1330Trpfs * 221) [57]. It is characterized by proportionate short stature (<5th percentile; males 141–12 cm and females 136–149 cm) with stocky appearance and severe progressive osteoarthritis of the large ones [58]. Radiographic features include endplate irregularity and vertebral body sclerosis with mild and variable epiphyseal changes associated with delayed bone age [59].
Cartilage oligomeric matrix protein
Cartilage oligomeric matrix protein (COMP) is a homopentameric protein belonging to the thrombospondin gene family (TSP), whose gene is located on chromosome 19p12-13.1 [60]. The structure of TSP proteins has a common organization consisting of an N-terminal domain, 4 type 2 epidermal growth factor (EGF)-like repeats, 7 calcium-binding repeats of type 3, and a C-terminal globular domain [61,62]. Fare clic o toccare qui per immettere il testo.
The function of COMP would seem to be stimulation of chondrogenesis and proliferation of chondrocytes [63] and mediation of the interaction between proteins of the ECM in cartilage and other tissues [64,65]. More than 50 mutations have been identified, and most of these have been found in highly conserved repeat type 3 calcium-binding domains [66].
COMP mutations would be responsible for the accumulation of COMP and other ECM proteins within the ER of chondrocytes, causing a decrease in the proliferation of chondrocytes. COMP mutations are associated with 2 autosomal dominant skeletal dysplasias: pseudoachondroplasia (PSACH), a severe condition of dwarfism, and MED, a milder short stature disorder [67]. While all PSACH are related to COMP mutations, only 66% of MED are caused by COMP mutations [68].
PSACH is a condition of disproportionate dwarfism involving both the spine and limbs associated with joint abnormalities [69]. PSACH patients initially show normal growth, but begin to show the first signs of short stature by the end of the first year. The result is an adult height equivalent to that of an average 6-to 8-year-old child [70]. The face traits are distinctive and characteristic features. All joints are affected and are extremely lax. Joint pain is a major complication that begins in childhood and persists into adulthood [71].
Characteristics include radiographic findings of long bones shortening, enlarged and irregular metaphyses, small underossified femoral epiphyses, and platyspondyly [71].
MED is the milder skeletal dysplasia, presenting with epiphyseal abnormalities and joint pain which start in childhood [72]. MED patients are diagnosed around 5 years of age due to abnormal gait and/or joint stiffness and pain. Height is only slightly altered, with some patients achieving normal height. Early osteoarthritis, which primarily affects the hips, requires hip replacement in early adulthood [66].
Matrilin-3
Matrilin-3 (MATN3) is an ECM protein that is part of the matrilin family, whose gene is located on chromosome 2p24-p23. The matrilin family consists of 4 proteins formed by 1 or 2 Von Willebrand factor A (VWFA) domains, a variable number of EGF-like domains, and an α-helical spiral domain. MATN3 contains a VWFA domain, 4 EGF domains, and a C-terminal spiral domain [73,74]. The matrilins participate in the ECM assembly, and MATN3 mediates the interactions between collagen fibrils and the other ECM proteins [66,75]. Furthermore, MATN3 seems to play a role in chondrogenesis, premature maturation of chondrocytes, and ossification. In MATN3 knockout mice, growth plate chondrocytes transform early into a prehypertrophic and hypertrophic phenotype and form an expanded zone of hypertrophy [76]. MED can also be caused by MATN3 mutations [77,78] which cause retention of the protein within the ER, resulting in cellular stress [79,80].
A disintegrin and metalloprotease with thrombospondin motifs 17
A disintegrin and metalloprotease with thrombospondin motifs 17 (ADAMTS17) belongs to the ADAMTS family of ECM proteases, functionally related to fibrillins. It would seem that ADAMTS17 promotes the secretion and/or deposition of FBN1 and type I collagen in the ECM [81,82]. Furthermore, both in cell culture systems and in vivo, direct interactions among fibrillin-1, ADAMTS17, and LTBP2 have been demonstrated.
All ADAMTS proteases are characterized by a similar structure; they have a conserved N-terminal domain and a variable C-terminal auxiliary domain, which is involved in binding with other ECM proteins [83].
Mutations in ADAMTS17 are associated with WMS. Individuals with WMS type IV due to ADAMTS17 mutations present with predominantly musculoskeletal and ocular manifestations, having less severe cardiac involvement [82,84,85].
In genome-wide studies, the involvement of the ADAMTS17 locus is often correlated with human height [86]. Therefore, it is possible that familial short stature may be associated with suspected monoallelic ADAMTS17 deficiency. However, this hypothesis needs further investigation.
Latent transforming growth factor beta-binding proteins
LTBPs are important ECM proteins; 4 isoforms of latent transforming growth factor beta-binding proteins (LTBP2) interacting with fibrillin microfibrils have been identified in the human genome [87]. Electron microscopic examination revealed that LTBP2 could play a role in the formation of elastic fibers and in the assembly of extracellular microfibrils [39].
LTBP2 interacts with heparin and heparan sulfate proteoglycans through its N-terminal domain and, more weakly, through its central region [88]. These interactions would lead to further anchoring of the microfibrils to basal membranes.
Homozygous mutations in the LTBP2 gene cause WMS type III, primary congenital glaucoma [89], and appear to be involved in the pathogenesis of Marfan syndrome [90].
Fibronectin 1
Fibronectin 1 (FN1) (2q35) encodes for fibronectin [91]. Mutations in the FN1 gene impair cysteine residues that form disulfide bonds, on which depends the 3-dimensional structure of the protein, and their perturbation leads to instability and possible risk of degradation by metalloproteinases (MMP9 and MMP13) [92].
Mutations in the N-terminal domain, which is necessary for fibronectin interaction to form fibrils, lower the number of fibrils in the cell matrix [93]. Some pathogenic variants in the III-2 domain result in greatly reduced secretion of the protein and accumulation of abnormal fibronectin within the cells. Molecular genetic testing may identify some heterozygous mutations associated with spondylometaphyseal dysplasia, corner fracture type (SMDCF), a skeletal dysplasia characterized by short stature which may be present at birth or develop in early childhood. Individuals may present with short limbs and/or short trunk, and the final adult height is more than 2 SD below the mean. Some distinguishing features reported in individuals with FN1-SMDCF include nonspecific dysmorphic facial features and frequent fractures due to low bone mineral density. In addition, limited joint mobility and chronic pain are common.
Radiological characteristics include enlarged metaphyses with corner fracture-like lesions, coxa vara, shortened long bones, scoliosis, and vertebral anomalies (e.g., ovoid-shaped vertebral bodies, anterior wedging, narrow intervertebral spaces, vertebral fusion, vertebral hypoplasia) and joint stiffness with pain [94].
Conclusions
Short stature is one of the main reasons for a referral to a pediatric endocrinologist. In this review, we discussed the main causes of short stature, focusing on single genes defects involving the ECM of the growth plate. These mutations most often cause rare, complex, and multisystemic clinical pictures characterized by severe harmonic or disharmonious short stature. However, we have also observed that some variants of these mutations are frequently recognized in patients with idiopathic short stature who do not have syndromic clinical features. The new molecular genetics techniques will make it possible to recognize an increasing number of new genes and gene variants implicated in short stature, thereby allowing for the recognition of genetic short stature, which still remains misdiagnosed and identified as idiopathic forms.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Writing - original draft: MAS, AQ, FC; Writing - review & editing: MASi, AQ, FC