Growth hormone deficiency in a boy with Wiedemann-Steiner syndrome: a case report and review
Article information

Highlights
· A Korean pediatric case of Wiedemann-Steiner syndrome (WSS) highlights a de novo KMT2A gene variant, presenting with facial dysmorphism and developmental delay. The patient, diagnosed with growth hormone deficiency (GHD), showed significant height improvement with recombinant human growth hormone treatment, emphasizing the need to consider WSS in GHD cases.
To the editor,
Wiedemann-Steiner syndrome (WSS) is a rare, autosomal dominant genetic disorder caused by mutations in the lysine (K)-specific methyltransferase 2A (KMT2A) gene [1,2]. It is characterized by a broad phenotypic spectrum, including facial dysmorphism, hypertrichosis, short stature, and developmental delay [1,2]. It was recently reported that growth hormone deficiency (GHD) might also be found in patients with WSS [3,4] Herein, we report a case of WSS in a boy who also had GHD.
A 2-year-old boy born to nonconsanguineous Korean parents presented with short stature and developmental delay. He was born at 39+2 weeks of gestational age with a birth weight of 3,595 g (50th–75th percentile), length of 50.5 cm (50th–75th percentile), and head circumference of 36.5 cm (90th–95th percentile). Polyhydramnios was apparent in the prenatal period, with no other specific perinatal or family history. His delivery was normal, with Apgar scores of 8 and 9 at 1 and 5 minutes, respectively. Neonatal screening tests, including tandem mass, were all normal. At 9 months of age, axial hypotonia and developmental delay were suspected. He was able to walk independently at 18 months of age. At 24 months of age, the BSID-II (Bayley Scales of Infant Development II) showed a global developmental delay. An examination revealed hypermetropia, strabismus, high-arched palate, open mouth, torticollis, and simian lines. Genetic studies, including multiplex ligation-dependent probe amplification, ruled out congenital myotonic dystrophy and other related disorders.
His growth velocity decreased gradually after 18 months. At 24 months, his height was 81.1 cm (below the 3rd percentile), weight was 10.5 kg (10th percentile), and head circumference was 48.6 cm (50th–75th percentile). His mean parental height was 173.5 cm (25th–50th percentile). His thyroid function (TSH 1.95 uIU/ml and FT4 1.54 ng/dL) was normal. The insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 levels were 32.4 ng/mL (10th–25th percentile) and 1,970 ng/mL (50th–75th percentile), respectively. The patient was diagnosed with GHD through a growth hormone (GH) provocation test (peak GH level was 6.9 ng/mL). Magnetic resonance imaging of the brain showed normal findings.
Because of the patient's dysmorphic features and developmental delay, whole-exome sequencing was performed and identified a de novo heterozygous variant of KMT2A:c.731T>G(p.Leu244*. This variant, which is located within exon 3 of the KMT2A gene on chromosome 11, was confirmed by targeted Sanger sequencing (Fig. 1). The variant creates a stop codon at codon 244, resulting in premature termination of the protein product and was classified as likely pathogenic according to the recommendations of the American College of Medical Genetics (ACMG) guidelines [5]. This novel variant was added to the Clinvar database (SCV002012176.1).

Family tree and chromatograms of the causative genetic mutation in the patient and parents. The blank square and circle indicate the unaffected father and mother, respectively; the filled square indicates the affected patient. Sanger sequencing confirmed a c.731T>G (p.Leu244*) mutation in the KMT2A gene of the patient. However, the parents did not have a mutation in the KMT2A gene. WT, wild type.
Recombinant human GH treatment was started with an initial dose of 0.1 IU/kg/day, which resulted in a significant improvement in height. The child's height increased to the 10th percentile after 1 year.
To date, more than 50 studies of WSS have been reported. According to the Leiden Open Variation Database (2022/03/28), 281 public variants have been reported. The Clinvar database lists 137 variants, with 102 classified as pathogenic or likely pathogenic (2022/03/24). In addition, 35 variants are identified as nonsense mutations, 50 as missense mutations, 41 as frameshift mutations, and 10 as splicing mutations. The characteristics and pathogenic variants of patients with WSS reported in the literature are presented in Table 1.
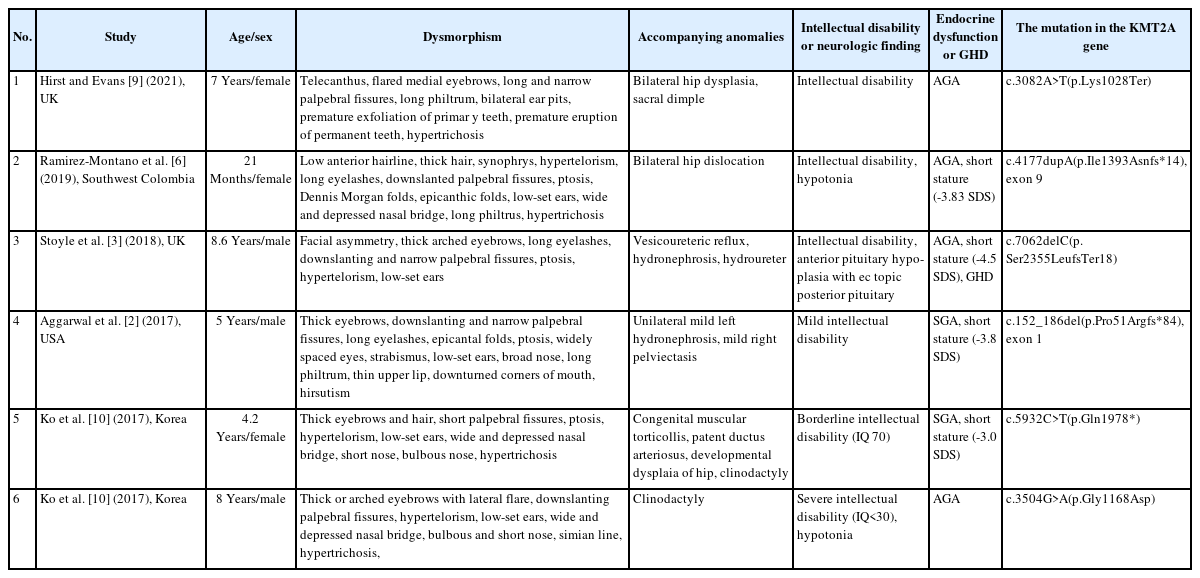
Summary of phenotypes and genotypes in patients with Wiedemann-Steiner syndrome reported in the literature
KMT2A, located on chromosome 11q23.3, contains 36 exons; however, most mutations are on exon 27, with one on exon 3 [2]. The KMT2A gene encodes a histone 3 lysine 4 methyltransferase, which is critical for DNA packing, chromatin modification, and gene expression regulation as an epigenetic writer. As KMT2A is widely expressed in almost all tissues, the phenotypes of patients with WSS involve multiple body systems, including facial features, skeletal anomalies, and developmental disability [6].
Recent studies have suggested a link between WSS and GHD, although the underlying mechanism remains unclear [3,4]. GHD has also been observed in other syndromes that involve the KMT2 gene family, such as Kabuki syndrome and CHARGE, which indicates a potential role in the hypothalamic pituitary axis [7,8].
In conclusion, this report details the first case of WSS in a Korean pediatric patient with accompanying GHD. Our case emphasizes the importance of considering WSS in children with GHD who present with other clinical features such as facial dysmorphism and developmental delay. This case also contributes valuable insights to the understanding of the phenotypic spectrum of WSS and its association with GHD.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics statement
Written informed consent for publication of this case report was obtained from the patient's parents.
Acknowledgements
We thank the patient and his parents for their cooperation.