Highlights
· Fibroblast growth factor receptor 3 (FGFR3)-related skeletal dysplasia is a relatively common disease entity. Early diagnosis is essential for timely management of complications. Emerging targeted therapies are expected to provide better outcomes in these patients in the near future.
Introduction
Fibroblast growth factors (FGFs) and FGF receptors (FGFRs) play essential roles in human axial and craniofacial skeletal development. FGFR3 inhibits chondrocyte proliferation and differentiation and promotes chondrocyte apoptosis [1]. Consequently, pathogenic variants of the FGFR3 gene, which mostly exhibit gain-of-function, are responsible for a group of chondrodysplasias characterized by short-limb dwarfism, macrocephaly, and a narrow thorax. Since the discovery of the activating FGFR3 variant as the genetic cause of achondroplasia (ACH) in 1994, other disorders caused by gain-of-function FGFR3 variants have been elucidated. These include hypochondroplasia (HCH), thanatophoric dysplasia types 1 (TD1) and 2 (TD2), and severe ACH with developmental delay and acanthosis nigricans (SADDAN) [2]. Camptodactyly, tall stature, scoliosis, and hearing loss (CATSHL) syndrome has been reported in 2 families, showing excessive skeletal growth due to loss-of-function of FGFR3 [3,4]. The appropriate diagnosis of FGFR3-related skeletal dysplasias depends on the combination of family history, clinical and radiologic findings, and molecular tests [5]. To date, treatment of skeletal dysplasias has been largely supportive, including skeletal surgery. However, in recent decades, several therapeutic strategies targeting the FGFR3 signaling pathway have been investigated. The first drug, vosoritide, was approved and is currently used in the United States and Europe [2,6,7]. In this review, we describe the clinico-radiologic and molecular findings of FGFR3-related skeletal dysplasia, and discuss recent therapeutic achievements, focusing on pharmacological approaches in ACH.
Pathophysiology of FGFR3-related skeletal dysplasia
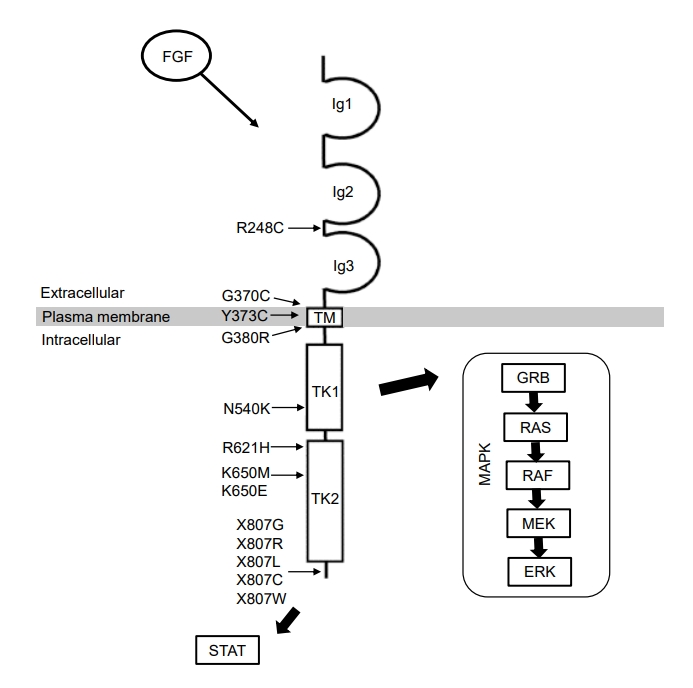
FGFR3 is a receptor tyrosine kinase that comprises an extracellular ligand-binding domain, a transmembrane domain, and 2 intracellular tyrosine kinase domains [8]. Binding of various FGFs to FGFR3 induces receptor dimerization and transphosphorylation of tyrosine residues [9], leading to activation of several downstream signaling pathways, including mitogen-activated protein kinase (MAPK) cascades (Fig. 1) [10,11]. Gain-of-function variants in the FGFR3 gene lead to increased signal transduction of FGFR3, thus suppressing chondrocyte proliferation and differentiation [12]. This impairs endochondral ossification of long bones, leading to poor extension of the long bones. Loss of FGFR3 function induces overgrowth of long bones in mice with knockout variants of FGFR3 and individuals with CATSHL syndrome [3,10,13,14], which supports the important role of FGFR3 as a negative regulator of bone growth.
Clinical characteristics and diagnosis
Diagnosis of FGFR3-related skeletal dysplasia is established based on clinical and radiological findings and/or identification of FGFR3 pathogenic variants (Table 1). Radiological evaluation with a complete skeletal survey plays a crucial role in the diagnostic workup of skeletal dysplasia. Radiographic survey includes the skull, spine, chest, pelvis, legs, arms, and hands (Table 2) [15].
1. Achondroplasia
ACH (MIM 100800) is the most common cause of short-limb dwarfism and affects more than 250,000 people worldwide [16]. Estimated prevalence ranges from 0.4–0.6 per 10,000 live births [17]. ACH follows an autosomal-dominant inheritance pattern, with more than 80% of cases attributable to spontaneous mutation [16]. The increased incidence of sporadic cases is partially associated with advanced paternal age [18]. Virtually all patients carry the 2 common pathogenic variants of the FGFR3 gene (c.1138G>A and c.1138G>C), which results in identical mutant proteins, p.Gly380Arg (G380R) [19]. The diagnosis can easily be made in early infancy based on physical and radiological features. The clinical features include rhizomelic shortening of the limbs, distinctive facial features (frontal bossing, depressed nasal bridge, and midface hypoplasia), macrocephaly, brachydactyly, and trident hands. Other reported associations include foramen magnum narrowing, ventricular enlargement, obstructive sleep apnea, a narrow thorax, upper airway stenosis, spinal stenosis, genu varum, limited elbow extension, and joint hyperextensibility (especially of the knees) [20-22]. In the United States, the mean adult height is approximately 131±5.6 cm for men and 124±5.9 cm for women [23]. Intelligence and lifespan are usually in the normal range, although brainstem compression increases the risk of unexpected infant death [24]. Specific features on radiographs include rhizomelic shortening of the long bones with metaphyseal flaring, shortened femoral necks, proximal femoral radiolucency, chevron shape of the distal femoral epiphyses, relatively long fibula bones compared to tibia bones, short and broad pelvis with horizontal acetabuli and squaring of iliac wings, and short pedicles of the vertebrae with caudal narrowing of the lumbar interpedicular distance [5,25]. Although molecular confirmation is not generally needed to diagnose ACH in individuals with typical findings, molecular analysis may aid in establishing a diagnosis in those with atypical presentations or receiving new treatments.
2. Hypochondroplasia
HCH (MIM 146000) is the mildest form of FGFR3-related dwarfism, and its expected prevalence is similar to that of ACH. HCH is inherited in an autosomal-dominant manner, with the majority of new cases caused by de novo variants in the FGFR3 gene, which have been associated with advanced paternal age [26]. Approximately 70% of patients are heterozygous for the 2 recurrent variants of FGFR3 (c.1620C>A and c.1620C>G), causing identical amino acid changes of p.Asn540Lys (N540K) in its tyrosine kinase domain [27-29]. HCH diagnosis is challenging because of the subtle clinico-radiologic features and phenotypic overlap with other skeletal dysplasias. There is a wide spectrum of clinical severities, and clinical features may become apparent over time. Patients usually present with short stature with mild limb shortening, macrocephaly, lumbar lordosis, and brachydactyly [30]. In South America, mean adult height was reported to be 143.6 cm (range, 131–154.5 cm) for men and 130.8 cm (range, 124–138 cm) for women [31]. Although the medical complications prevalent in ACH (e.g., spinal stenosis) are less frequent [32], intellectual disabilities and seizures may be more common in HCH [33,34]. The skeletal features of HCH are similar to those in ACH but are much milder. Because there is no consensus on the clinical diagnosis of HCH, molecular confirmation is necessary to distinguish HCH from other skeletal dysplasias in patients with overlapping phenotypes.
3. Thanatophoric dysplasia
TD is the most common lethal skeletal dysplasia, with a reported incidence of 0.2 to 0.5 per 10,000 live births [35]. TD is typically caused by de novo pathogenic FGFR3 variants, which have been associated with an advanced paternal age effect [36]. The clinical and radiographic features of TD are evident prenatally or immediately after birth. These include micromelia, macrocephaly, distinctive facial features (frontal bossing and a depressed nasal bridge), and a narrow thorax. There are 2 clinical types of TD: type 1 (TD1, MIM 187600) and type 2 (TD2, MIM 187601). The characteristic bowed femurs in TD1 may distinguish it from TD2. TD2 is characterized by severe craniosynostosis with cloverleaf skull deformity, which is rarely seen in TD1 [37]. Most affected individuals die in the perinatal period from respiratory failure due to narrow thorax, lung hypoplasia, and/or compression of the brainstem due to narrowing of the foramen magnum [38]. Neonates typically require long-term respiratory support to survive. Rare long-term survivors have been reported, who are often ventilatordependent, with marked growth deficiency and extreme intellectual disability [39-44].
4. SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans)
SADDAN (MIM 616482) is a rare skeletal dysplasia caused by a heterozygous FGFR3 pathogenic variant (c.1949A>T) that results in amino acid changes in p.Lys650Met (K650M) [45,46]. The clinical features include extremely short stature, macrocephaly, characteristic facial features (frontal bossing, depressed nasal bridge, and midface hypoplasia), rhizomelic or mesomelic limb shortening, severe tibial bowing, seizures, developmental delay, and acanthosis nigricans. The clinical features overlap with those of TD, however, individuals with SADDAN often survive beyond infancy without ventilatory support. However, early mortality with respiratory failure has been reported, and respiratory support may be needed during the neonatal period [45-47].
5. CATSHL (camptodactyly, tall stature, scoliosis, and hearing loss) syndrome
CATSHL syndrome (MIM 610474) is a distinct skeletal dysplasia caused by loss of FGFR3 function [3]. Since its first description in 1996, 2 families with CATSHL syndrome have been reported, one with a heterozygous variant and the other with a homozygous variant [3,4]. The c.1915G>A variant in the FGFR3 gene results in an amino acid change in p.Arg621His (p.R621H) that induces loss-of-function of the gene. Major features included camptodactyly, tall stature (mean adult height of 195.6 cm in men and 177.8 cm in women), and hearing loss. Other observed findings included kyphoscoliosis, a high palate, microcephaly, and intellectual disabilities.
Management
Management of skeletal dysplasia is currently largely supportive, including periodic measurements of growth, surgery for craniocervical junction stenosis and hydrocephalus, management of middle ear infections and obstructive sleep apnea, and surgical intervention for spinal stenosis, thoracolumbar kyphosis, and leg bowing [22]. Other treatment interventions may include antiseizure medications, hearing aids, and individualized developmental support. Treatment for short stature depends on parental concerns and expectations. Recombinant growth hormone (GH) does not appear to be effective in ACH, with an expected additional adult height of only about 3 cm (approved only in Japan) [48]. On the other hand, a meta-analysis of 113 children with HCH revealed a greater response to GH therapy in children with HCH with major catch-up growth after 12 months, and improvement in height remained constant until 36 months of GH treatment [49]. However, the ultimate success of GH treatment in HCH remains uncertain due to the lack of data on final adult height in GH-treated patients. Extended limb lengthening is controversial in both ACH and HCH because of the frequent and often serious complications [50]. It is recommended that this invasive procedure be postponed until the patient can make an informed decision. In 2021, the FDA approved a C-type natriuretic peptide (CNP) analog, vosoritide, to increase height in ACH individuals aged ≥5 years whose epiphyses are not closed [51].
Emerging therapies targeting the FGFR3 signaling pathway
Cancer biology has inspired attempts to block FGFR3 because aberrant FGFR signaling is also implicated in various cancers, including bladder and cervical cancers [52]. Therapeutic strategies targeting bone tissue affected by gain-of-function FGFR3 variants inhibit various steps of the FGFR3 pathway, targeting the FGF ligand or the FGFR3 receptor, tyrosine kinase activity of FGFR3, and downstream signals. Several clinical trials are currently underway for FGFR3-related skeletal dysplasias, mainly for ACH (Table 3).
1. CNP analog (vosoritide)
CNP is expressed in proliferating and prehypertrophic chondrocytes and stimulates chondrocyte proliferation by inhibiting MAPK signaling at the RAF-1 level [53-55]. A randomized, double-blind, phase-III trial of vosoritide (ClinicalTrials.gov identifier: NCT03197766) demonstrated an average increase of 1.6 cm/yr in annual growth velocity in up to 121 children with ACH aged 5 to <18 years [56]. Children who completed this trial will be followed to assess long-term efficacy and safety (ClinicalTrials.gov number, NCT03424018). Currently, vosoritide (Voxzogo; BioMarin, San Rafael, CA, USA) is available for patients with ACH in Europe and the United States. Additionally, a phase-II, randomized, double-blind trial of vosoritide in infants and younger children with ACH (age range, 0 to <60 months) (ClinicalTrials.gov identifier: NCT03583697) was recently completed (results not yet published). A phase-II, randomized, open-label trial evaluating the safety of vosoritide in infants at risk for cervicomedullary decompression surgery is now recruiting participants aged 0–12 months (ClinicalTrials. gov identifier: NCT04554940). Another phase-II study of vosoritide (ClinicalTrials.gov identifier: NCT04219007) is ongoing in children 3–10 years of age with selected genetic causes of short stature, including HCH.
2. TransCon CNP
Another CNP derivative with a longer half-life that can be administered by weekly subcutaneous injection, has also been developed (TransCon CNP; Ascendis Pharma A/S, Hellerup, Denmark). A phase-I study has been completed, and an observational study to document the natural history is recruiting infants and children with ACH aged 0–8 years (ClinicalTrials. gov number, NCT03875534; ACHieve). Currently, children aged 2–10 years with ACH are being enrolled in phase-II, multicenter, randomized, placebo-controlled, dose-escalation trials to evaluate the safety and efficacy of TransCon CNP (ClinicalTrials.gov number, NCT04085523; ACcomplisH, and NCT05246033; ACcomplisH China).
3. Soluble FGFR3 isoform as a decoy receptor (TA-46/Recifercept)
A soluble isoform of FGFR3 (TA-46, now called Recifercept) is currently in clinical development to treat children with ACH. It is composed of the extracellular domain of FGFR3 and acts as a decoy receptor, which prevents FGF from binding to the mutated FGFR3 by competing for physiologic ligands (i.e., FGFs 9, 18). A phase-I study using Recifercept (Pfizer Ltd., New York, NY, USA) has been completed, and an observational study investigating baseline clinical characteristics of children with ACH aged 0–10 years (ClinicalTrials.gov number, NCT03794609) has been conducted. Currently, 2 phase-II studies to document the safety, tolerability, and effectiveness of Recifercept are recruiting children with ACH aged 3 months to 10 years (ClinicalTrials.gov number, NCT04638153), and aged 15 months to 12 years (ClinicalTrials.gov number, NCT05116046).
4. Tyrosine kinase inhibitor (NVP-BGJ398/Infigratinib)
Infigratinib (NVP-BGJ398), an orally bioavailable and selective FGFR1-3 selective tyrosine kinase inhibitor, counteracts the hyperactivity of FGFR3 by inhibiting phosphorylation of FGFR, resulting in attenuation of its downstream signaling [57]. Phase-I studies using this tyrosine kinase inhibitor (infigratinib; QED therapeutics) have been completed, and an observational study to evaluate its natural history has commenced in children with ACH aged 30 months to 10 years (ClinicalTrials.gov number, NCT04035811; PROPEL). A phase-II study evaluating the safety and efficacy of infigratinib (ClinicalTrials.gov number, NCT04265651; PROPEL 2) is recruiting children with ACH aged 3–11 years. Additionally, a phase-II, multicenter, open-label extension (OLE) study (ClinicalTrials.gov number, NCT05145010; PROPEL OLE) is ongoing to evaluate the long-term safety and efficacy of infigratinib in children aged 3–18 years with ACH.
5. Drug repurposing: meclozine and statins
Meclozine, an antihistamine that has long been used to treat motion sickness, was considered a putative treatment because it downregulates phosphorylation of ERK in the MAPK pathway. In a preclinical study, oral administration of meclozine significantly increased the length of long bones in transgenic ACH mouse pups [58]. Additionally, a class of cholesterol-lowering drugs, statins, rescued cartilage degradation in patient-derived induced pluripotent stem cell models and led to a significant recovery of bone growth in transgenic ACH mouse pups [59]. However, further experiments beyond animal models have not been conducted on either meclozine and statins.
Conclusions
FGFR3-related skeletal dysplasias are a dynamic group of disorders with evolving skeletal phenotypes throughout the lives of affected individuals. Although some disorders are perinatally lethal, others have a relatively normal life expectancy. Early diagnosis is crucial for appropriate genetic counseling and prevention of serious medical complications through multidisciplinary care. With the advent of several therapeutic options targeting FGFR3 in ACH, these new therapies are expected to be effective in other FGFR3-related skeletal dysplasias. Hopefully, they will provide better, nonsurgical options for these patients to improve their quality of life and minimize medical complications.