Endocrinal dysfunction in children with Down syndrome
Article information

Abstract
Down syndrome (DS) is the most common genetic disorder in live-born infants. Children with DS are at increased risk of numerous endocrinal comorbidities. The information contained in this article will provide pediatricians with a narrative overview of different presentations, diagnoses, and management recommendations of various endocrinal disorders in children with DS. We systematically searched PubMed, Embase, Google Scholar, MEDLINE, EBSCO, and Science Direct, and potentially relevant articles were identified and retrieved from electronic and print journals.
Highlights
Down syndrome (DS) is the most common chromosomal condition. Children with DS have a higher chance of developing endocrine disorders such as thyroid dysfunction, diabetes mellitus, obesity, short stature, vitamin D deficiency, low bone mineral density, and gonadal dysfunction than the general population. Pediatric endocrinologists should be aware of the management of endocrine problems that can occur in children with DS.
Introduction
Down syndrome (DS), caused by the presence of a third copy of chromosome 21, is the most frequently occurring chromosomal condition, affecting from 1 in 700 to 1 in 1,500 live-born babies [1]. DS is associated with developmental disabilities as well as medical diseases such as congenital heart disease, pulmonary abnormalities, sleep-related breathing disorders, and endocrinal dysfunction [2]. Children with DS have a higher likelihood of developing endocrine disorders such as thyroid dysfunction, diabetes mellitus, short stature, vitamin D deficiency, and obesity than does the general population (Table1) [3]. Precise diagnostic modalities and effective management for these disorders do exist; however, best practices for some of these endocrine abnormalities have not yet been confirmed [4]. This review will discuss the characteristics of the different endocrine disorders in children with DS and contribute updated management recommendations.
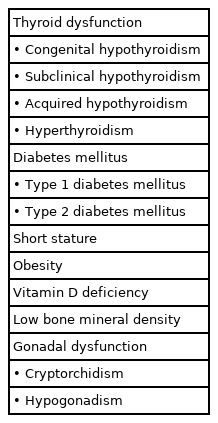
Endocrinal dysfunctions in children with Down syndrome
Thyroid dysfunction
Thyroid dysfunction is the most common endocrine abnormality in DS children. Compared to the general population, thyroid dysfunction is 25–38 fold more likely in the DS population [5,6]. The thyroid disorders that occur in children with DS include congenital hypothyroidism (CH), subclinical hypothyroidism (SH), acquired hypothyroidism, and hyperthyroidism [7]. Thyroid function tests (TFTs) like thyroid-stimulating hormone (TSH), free T4 (FT4), and free T3 (FT3) are routinely used for assessment of thyroid dysfunction [8]. TSH is a very sensitive marker of changes in thyroid status; incremental alterations in FT4 level lead to logarithmic changes in the secretion of pituitary TSH [9]. Measurement of both TSH and FT4 is advised to determine thyroid function accurately and to monitor treatment in children with thyroid disorders [8]. Screening using TFT for thyroid disease at birth, at 6 months of age, at 12 months, and annually thereafter is the standard of care [10-13].
Congenital hypothyroidism
The reported incidence of persistent primary CH in DS is much higher than in the general population, varying between 1:113 and 1:141 live births in DS versus 1 in 2,000 to 3,000 live births in the general population [14], with one study estimating it to be 28 times higher than in the general population [5]. The etiology of CH does not seem to be thyroid agenesis, as most patients have no abnormalities in thyroid scans [15]. Reported causes of CH are thyroid hypoplasia, thyroid ectopia, or partial agenesis [16]. Studies on the pathogenesis of CH in children with DS have suggested the following hypotheses: (1) delayed maturation of the hypothalamic-pituitary-thyroid axis leading to higher TSH level with normal FT4 and FT3 levels, (2) peripheral resistance to thyroid hormones leading to inappropriate TSH secretion and release due to a central disorder, and (3) TSH insensitivity and reduced TSH bioactivity [17,18]. Children with DS with CH are at increased risk of developing congenital anomalies, especially congenital heart diseases and gastrointestinal anomalies, compared to patients with DS and without CH [19].
Subclinical hypothyroidism
SH is defined as serum TSH concentration above the upper limit of the reference range in the presence of normal thyroid hormone levels [9]. SH is the most common thyroid abnormality in children with DS, with a prevalence that varies between 7% and 40% [20]. Hashimoto thyroiditis (HT) is the most common etiology of SH in DS children, and HT is more likely to be present with SH in DS compared to the control population [21]. SH is usually asymptomatic and tends to be transient and self-limiting in children with DS [22]. However, some children display mild symptoms such as hypotonia or weight gain, although these symptoms often occur in children with DS and are not sufficient for diagnosis [23]. Ultrasound scans show normal thyroid gland in the majority of cases [9]. Controversy exists over whether to treat DS children with SH, also over the cutoff point to use for the decision to treat [9,21,22]. Many authors have recommended not treating SH in children with DS because of the benign and remitting nature of the condition [23-26]. On the other hand, other authors argue that early thyroxin treatment is potentially harmless and can improve growth, motor development, and intellectual function in children with DS, a population with delayed development [27-29]. From consideration of all data mentioned above, it has been suggested that treatment of SH should be confined to DS children who progress to overt hypothyroidism (OH) on follow-up and to those with TSH >10 μU/mL with the existence of goiter or the presence of antithyroid antibodies [30].
Acquired hypothyroidism
HT is the most common cause of acquired hypothyroidism in children with DS [26]. Around 10% of school-aged children with DS have OH, which is defined as elevated TSH level combined with low FT4. The prevalence of OH increases with age and presence of antithyroid peroxidase (TPO Ab) [31]. The increased incidence of autoimmune disease in DS has been hypothesized to be attributable to the following factors: (1) altered immune function in DS, either humeral or cellular, (2) mutations in the autoimmune regulator (AIRE) gene located in the 21q22.3 region involved in immune regulation, (3) alterations in the regulation of pro- and anti-inflammatory cytokines, (4) the suppressive effect of interferon-alpha and its toxic effect on the thyroid gland, and lastly, (5) an association with the DQA1 0301 allele linked to the increased association of autoimmune thyroid disease and celiac disease [30]. Diagnosis of OH on the basis of clinical background is not definitive as symptoms and signs might not be clear or might be dismissed as part of the DS clinical criteria. So, biochemical diagnosis with TFT is essential [7]. In contrast to the general population, autoimmune hypothyroidism in DS is characterized by the following features: equal gender distribution, lower age at diagnosis, lower frequency of positive family history of thyroid disease, lower antibody titer at diagnosis, and increased association with other autoimmune disorders [32]. OH requires treatment with thyroid hormone in addition to regular monitoring of TFT [28].
Hyperthyroidism
Graves' disease (GD) is the main etiology of hyperthyroidism in DS, and its prevalence has been reported to be clearly higher in DS children and adolescents (0.66) than in the general population (0.02%) [33]. Moreover, GD in DS has no gender predominance unlike in the general population [34]. GD commonly presents during the adolescent period, is usually symptomatic, is easily diagnosed, and is also commonly associated with other autoimmune diseases [35]. Antithyroid drugs (ATDs) such as carbimazole and, rarely, surgery are options for management of GD in children with DS. However, the course of GD in DS children was usually mild and was well controlled with low-dose ATDs; moreover, some patients experienced remission [30].
Diabetes mellitus
Children with DS have a higher prevalence of diabetes mellitus (DM) than does the general population. Either type of DM (type 1 [T1DM] or type 2 [T2DM]) can appear in children with DS [36,37].
1. Type 1 DM
T1DM can affect up to 2% of DS children. There is a 4-fold increased risk of development of T1DM in DS children com pared to the general population of similar age [38]. One study reported that DS patients developed T1DM earlier than the general population, with a peak development time of around 8 years, compared to 14 years in the general population [39]. In another study, 22% of DS children developed T1DM before the age of 2 years compared to only 7% of children from the general population [40]. The etiology of T1DM in DS seems mainly due to increased subclinical islet autoimmunity and a lower frequency of the high-risk HLA genes compared to the general population [41]. Two studies reported increased rates of diabetes-associated autoantibodies in individuals with DS compared with the normal population without the expected increase in diabetes-associated HLA genotypes [42,43]. Other factors including mutations in the AIRE gene, located on chromosome 21 (21q22.3 region) and which regulates T-cell function and self-recognition, might result in autoimmunity and type 1 DM [44]. T1DM children with DS usually have better metabolic control despite lower insulin doses and lower rates of diabetesrelated complications [45]. Moreover, T1DM in DS is commonly linked with other autoimmune diseases, mainly autoimmune thyroiditis and celiac disease [38].
2. Type 2 DM
Studies revealed that the prevalence of T2DM in a pediatric population with DS ranged between 0%–3.6% [46]. However, its incidence rises with age, body mass index (BMI), family history, and being female [47]. A direct link has been established between DS and other metabolic diseases, especially T2DM and obesity, and a sedentary lifestyle [48]. Peripheral insulin resistance and declining β-cell function are the main factors in the development of T2DM in children with DS [49]. Treatment includes lifestyle modification, weight loss, and possible oral medication [48].
Short stature
Short stature is considered a characteristic feature of DS at all ages. 50) Newborns with DS have lower birth length, weight, and smaller head circumference compared with control newborns [51]. Height continues to be low up to puberty. Moreover, the growth velocity is markedly reduced during the normal period of accelerated growth during adolescence [52]. The causes of short stature in children with DS are open for debate; it might be related to deficiency of growth hormone (GH) secondary to hypothalamic or pituitary dysfunction [53]. However, some studies reported a deficiency of insulin-like growth factor 1 [54]. Other diseases such as thyroid dysfunction, celiac disease, obstructive sleep apnea, heart disease, and feeding difficulties can aggravate growth retardation [55]. DS-specific growth charts are currently available for assessment of stature in children with DS [50]. Some studies reported that GH treatment increases height and head circumference and improves psychomotor development in DS children [56,57]. However, a different report suggested that GH therapy is not recommended for patients with DS because it increases the risk of leukemia [58]. Further, GH is not approved by the U.S. Food and Drug Administration or the European Medicines Agency for treatment of short stature in children with DS [59].
Obesity
Children with DS are more likely to be overweight or obese than the general population of children without DS. The combined prevalence of overweight and obesity varied between studies from 23% to 70% [60]. However, studies indicate that obesity alone may be detected in around 7%–23% of children with DS [61]. It is hard to detect the exact factors leading to an increased risk of obesity in children with DS. However, it might be attributed to decreased resting energy expenditure, increased leptin, decreased metabolic rate and physical activity, unhealthy eating habits, and endocrine diseases, e.g., hypothyroidism, that have been reported in children with DS [62]. Obesity in children with DS might be associated with its resultant complications, including obstructive sleep apnea, hyperinsulinemia, dyslipidemia, T2DM, and gait abnormalities [63]. Clinical evaluation and monitoring of BMI are advised in children with DS beginning at 2 years of age [64]. Regular guidance and support for multifactorial strategies regarding healthy eating habits, physical activity, and avoidance of sedentary lifestyle should be implemented to reduce the incidence of overweight and obese children with DS [61].
Vitamin D deficiency
Children with DS are at increased risk of developing vitamin D deficiency or insufficiency with multifactorial etiology including inadequate exposure to the sun, inadequate vitamin D intake, malabsorption associated with celiac disease, or increased breakdown of vitamin D that accompanies anticonvulsant therapy [65]. Therefore, children with DS may require higher vitamin D supplementation than the recommended dietary allowance of 400 IU daily [66].
Low bone mineral density
Studies revealed that children with DS have lower bone mineral density (BMD), especially in the lumbar spine com pared to healthy individuals [67,68]. Other studies have suggested that bone appears to be produced at an abnormal rate during childhood in DS [69], and that the low bone density is most exaggerated in young adults [70]. Low activity levels, dietary insufficiency of vitamin D and calcium, insufficient exposure to the sun, and prolonged use of anticonvulsants can contribute to low BMD in children with DS. Moreover, endocrine disorders (hypothyroidism, hypogonadism) and autoimmune diseases that are commonly associated with DS can contribute to decrements in skeletal maturation and to bone-mass [71]. Management options include supplementation with calcium and vitamin D and an exercise program. Such treatment led to improvement in BMD in children with DS. However, bisphosphonates were suggested not to benefit patients with DS, as they decrease bone formation at baseline [67].
Gonadal dysfunction
1. Cryptorchidism
Studies reported a high incidence of cryptorchidism in DS children [72]. One study reported an incidence of 6.52%, with 4.35% being ascending or acquired undescended testes [73]. It is suggested that cryptorchidism in DS patients is due to failure of normal growth of the spermatic cord and/or mutation in the gene called insulin-like factor 3 and its receptor and/or fetal hormone deficiency [74]. Since there is a high incidence of tumorigenesis in DS cases, cryptorchid testes might be associated with a higher risk of testicular malignancy, which might have an early onset and poor prognosis [75]. Therefore, children with DS associated with cryptorchidism should be referred earlier for surgical descent of testis and appointed for regular follow-up for early detection of malignancy [76].
2. Hypogonadism
Gonadal insufficiency is a well-known feature of DS [50]. Previous reports indicated that male patients with DS can have hypogonadotropic hypogonadism characterized by increased levels of follicle-stimulating hormone and luteinizing hormone, presence in the infantile period, and progress throughout late puberty to adulthood. This condition can be attributed to dysfunction of both Leydig and Sertoli cells [66,77]. Moreover, studies have also reported that DS patients have lower total testosterone compared with the control population, which might be attributed to Leydig cell dysfunction due to the excess copy of the genetic material of chromosome 21 [78]. Furthermore, obesity associated with DS can be accompanied by increased aromatase activity, which converts testosterone to estradiol [79]. Regarding girls with DS, the reported abnormalities include delay in either adrenarche or menarche and hypogonadism [80].
Conclusions
DS is associated with multiple endocrine dysfunctions, particularly thyroid disease, DM, obesity, and short stature. This review gives useful information about endocrine dysfunctions in children with DS, which might help to optimize their management by effective interventions and facilitate attention to the clinical and biological outcomes of this special population.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: KM, HF; Data curation: KM, HF; Methodology: KM; Visualization: KM; Writing - original draft: KM, HF; Writing - review & editing: KM, HF