Genotype-phenotype correlations and long-term efficacy of pamidronate therapy in patients with osteogenesis imperfecta
Article information

Abstract
Purpose
Osteogenesis imperfecta (OI) is a rare bone fragility disorder caused by defects in type 1 collagen biosynthesis. This study investigated the genotype-phenotype correlations and the efficacy of pamidronate therapy in patients with OI in a single academic center.
Methods
This study included 24 patients with OI. A clinical scoring system was used to evaluate disorder severity. COL1A1 and COL1A2 genes were analyzed in 13 patients using Sanger sequencing. Genotype-phenotype correlations and the efficacy of pamidronate therapy were analyzed through a retrospective medical chart review.
Results
Of the 24 patients, 18 (75%) were classified as type I (12 with type Ia and 6 with type Ib), 2 as type III (8.4%), and 4 as type IV (16.7%). Type Ia patients showed relatively higher lumbar bone mineral density (BMD) standard deviation scores (SDS) and lower clinical scores than those with other types. Seven patients with qualitative mutations had lower lumbar BMD-SDS (P=0.015) and higher clinical scores (P=0.008) than 6 patients with quantitative mutations. The annual fracture frequency and lumbar BMD-SDS improved in patients with qualitative mutations after pamidronate treatment.
Conclusions
This study demonstrated that OI patients with qualitative mutations in COL1A1/2 had a more severe phenotype than those with quantitative mutations. Patients with qualitative mutations showed a significant reduction in fracture frequency and an increase in lumbar BMD-SDS after pamidronate treatment. Clinical score and genotype might be helpful for predicting phenotype and response to pamidronate therapy in OI patients.
Highlights
·OI is caused by heterozygous variants of genes encoding the α-helical chain of type 1 collagen.
·OI patients with qualitative COL1A1/2 mutations showed more severe phenotype than with quantitative mutations.
·Genotype is useful in predicting phenotype and response to treatment in OI patients.
Introduction
Osteogenesis imperfecta (OI) is a rare inherited bone fragility disorder caused by defects in type 1 collagen biosynthesis that results in skeletal phenotypes ranging from subclinical to lethal [1]. The incidence is estimated to be 1/15,000–20,000 births [2]. OI is classified according to the Sillence criteria based on inheritance patterns and clinical features such as presence of skeletal deformities, blue sclera, dentinogenesis imperfecta (DI), and hearing loss [3]. OI types I–IV are inherited in an autosomal dominant pattern. Type I (Ia if not accompanied by DI, and Ib if accompanied by DI) has the mildest severity, type II is associated with neonatal lethality, type III is characterized by severe progressive bone deformity, and type IV presents with moderately severe bone deformity. However, Sillence classification cannot reflect the clinical and genetic heterogeneities of the syndrome. With advances in molecular biolog y, OI categories have been expanded according to comprehensive radiographic, bone morphologic features, and molecular etiology, and now include type XX [4]. These classification systems have limitations related to variable extraskeletal features, such as hearing impairment, blue sclera, DI, and wormian bones [5]. To assess clinical severity in different types of OI, a clinical scoring system has been proposed [6].
OI is caused by mutations in collagen structural genes that result in changes in the structure of mature type 1 procollagen molecules, in protein amount, or in both [7]. Two α1-chains and one α2-chain constitute type 1 collagen, subsequently forming a rigid triple helix structure [8]. The helical domains contain highly repetitive amino acid sequences with glycine (Gly), which is located at the center of the triple helix in a Gly-X-Y sequence in each of the 3 α-chains, where X and Y can be any amino acid [9]. They subsequently fold in a zipper-like fashion into a triple helix in the lumen of the endoplasmic reticulum. Procollagen is then excreted into the extracellular space by osteoblasts and converted into collagen by cleaving the propeptide. Attachment of hydroxy groups and sugar molecules in the α-chains is a time-dependent process involving various enzymes [8].
Around 90% of OI cases are caused by autosomal dominant mutations in the COL1A1 and COL1A2 genes, which encode the α1- and α2-chains, respectively [8]. The genetic variants in COL1A1 and COL1A2 are categorized into 2 groups. One group consists of quantitative mutations, which mainly cause a decrease in the amount of type 1 collagen. These are mainly frameshift or nonsense mutations. The other group includes qualitative mutations, which results from amino acid substitutions that contain Gly peptide residues along the polypeptide chain, leading to production of structurally abnormal or less effective collagen. Quantitative mutations cause milder bone fragility than qualitative mutations. OI type II with neonatal lethality is exclusively associated with qualitative mutations [9]. Autosomal recessive forms of OI are caused by mutations in genes involved in mineralization defects (SEPINF1), posttranslational processing/modifications (CRTAP, LEPRE1, PIB, SERPINH1, BMP1, and TMEM38B), and folding/cross-linking of type 1 collagen (SERPINH1, FKBP10, and PLOD2), as well as in osteoblast differentiation (SP7 and WNT1) and function (WNT1) [4,10-15].
In Korea, few studies have investigated the genotype-phenotype correlations for OI and the short-term efficacy of pamidronate therapy, and only in a limited number of patients [16,17]. These studies have used the Sillence classification system to evaluate genotype-phenotype correlations. This study aimed to investigate the clinical characteristics and genotype-phenotype correlations in OI patients using a clinical scoring system and to assess the long-term efficacy and safety of pamidronate therapy [6].
Materials and methods
1. Subjects
This study included 24 patients (14 males and 10 females) with OI who were tre ated in a single academic center between January 1995 and December 2020. The patients were categorized into OI types according to the Sillence classification system [3].
Patient medical records were retrospectively reviewed, and the following variables were analyzed: age at diagnosis, height, weight, blue sclera, DI, hearing loss, increased ligament and skin laxity, scoliosis, long bone deformities, fracture frequency, and x-ray findings such as fractures at various healing stages, wormian bones, and codfish vertebrae. Diagnosis was based on clinical features and characteristic radiographic findings. Height and weight were expressed as standard deviation score (SDS) according to the standard references for Korean children [18].
The bone mineral density (BMD) of the lumbar spine and femoral neck was measured using dual-energy x-ray absorptiometry (Lunar Corp., Madison, WI, USA). The BMD results were adjusted for the Hologic system using the following formulas: "lumbar spine BMD according to the Hologic system=0.918×lumbar spine BMD according to the Lunar system – 0.038," and "femoral neck BMD according to the Hologic system=0.8638×femoral neck BMD according to the Lunar system – 0.039." BMD-SDS was obtained using age- and sex-matched references for Korean children [19,20].
A clinical scoring system was used to assess the severity of OI. The scoring system included 5 major criteria: annual fracture frequency, motor milestones, long bone deformities, heightSDS, and BMD-SDS. Each criterion was scored on a scale from 1 to 4, with 4 representing the greatest severity. The sum of all categories was classified as mild (5–10 points), moderate (11–15 points), and severe (16–20 points) (Supplementary Table 1) [6].
2. Pamidronate treatment of patients with OI
Pamidronate was indicated in patients with OI whose initial BMD-SDS was less than -2.0 and who experienced multiple long bone fractures or vertebral compression fractures. Pamidronate was administered intravenously at doses of 1 mg/kg over 4 hours for 3 consecutive days every 4 months [21]. The patients also received calcium carbonate (600 mg of elemental calcium per day) and 400 units of 25-dihydroxycholecalciferol per day. Biochemical profiles, including serum calcium, phosphorus, alkaline phosphatase, osteocalcin, and intact parathyroid hormone (PTH) levels, were assessed every 4 months. Eleven patients with OI previously reported by our group were included to evaluate long-term efficacy and safety of pamidronate therapy in our cohort [17].
3. Molecular analysis of the COL1A1 and COL1A2 genes
Mutation analysis was performed on 13 patients who consented to Sanger sequencing of their COL1A1 and COL1A2 genes using genomic DNA extracted from peripheral blood leukocytes. All 51 exons and exon-intron boundaries of COL1A1 and 52 exons and exon-intron boundaries of COL1A2 were amplified by polymerase chain reaction (PCR) using primers designed using Primer3 version 3.0 (http://bioinfo.ut.ee/primer3-0.4.0/). PCR products were directly sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit and an ABI3130x1 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
The pathogenicity of sequence variants was evaluated according to the standards and guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 22) In silico analyses of the sequence variants were conducted using prediction software including Polymorphism Phenotyping v2 (Polyphen-2; http://genetics.bwh.har vard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), Sorting Intolerant from Tolerant (https://sift.bii.a-star.edu.sg/), and Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu/). Mutations causing stop codons or frameshifts were classified as quantitative mutations, while mutations inducing amino acid substitutions were considered qualitative mutations.
4. Statistical analysis
Data were presented as mean±standard deviation. Statistical analyses were performed using IBM SPSS Statistics ver. 21.0 (IBM Co., Armonk, NY, USA). The Kruskal-Wallis test was used to compare parameters of each OI type, and the Mann-Whitney U-test was used to compare parameters between the patients with quantitative and qualitative mutations. The Wilcoxon signed-rank test was used to assess the efficacy of pamidronate therapy. Additionally, Fisher exact test was used to confirm genotype-phenotype associations. P-values of <0.05 were considered statistically significant.
Results
1. Clinical characteristics of patients with OI
Among the 24 OI patients, type I was the most common (n=18, 75%; 12 with type Ia and 6 with type Ib), followed by type IV (n=4, 16.7%), and type III (n=2, 8.4%). The median age at diagnosis was 5.9 years (range, 0.1–51.5 years). Patients were followed up for 14.3±7.3 years (range, 2.3–25 years). The patients' current median age was 22.7 years (range, 7.3–58.2 years). Type Ia patients had the highest mean height-SDS (-1.1±0.9) and type III patients had the lowest (-3.9±4.0, P=0.287). Likewise, type Ia patients had the highest mean height-SDS (-1.2±1.0) at the last follow-up, and type III patients had the lowest (-4.7±2.9, P=0.06). Blue sclera was a common phenotype in type Ia (11 of 12, 91.7%). Hearing loss was observed in type Ib (3 of 6, 50%) and type IV patients (2 of 4, 50%). Scoliosis was observed in 6 of the 18 (33.3%) type I patients and 4 of the 6 (66.7%) type Ib patients, and in all type III and type IV patients. Skeletal deformities, such as postfracture deformities, bowing of the tibia, and scoliosis, were present in all type III and type IV patients. Type Ia patients had significantly higher lumbar BMD-SDS than type Ib, type III, and type IV patients (P=0.022). Conversely, femur BMD-SDS did not differ significantly between type Ia, type Ib, type III, and type IV patients (P=0.785) (Table 1).

Demographic and clinical characteristics of 24 patients with osteogenesis imperfecta categorized according to the Sillence classification
According to the clinical scores, 15 patients were categorized into the mild severity group (7.8±1.7), 8 were categorized into the moderate severity group (12.3±1.4), and one type III patient with highest fracture frequency was classified as severe (17). In the mild severity group, type I patients were the most common (n=12, 80%, especially type Ia [11 of 15, 73.3%]), followed by 2 type IV patients (13.3%). In the moderate severity group, type I was the most common (n=5, 62.5%), with type Ib being the most common (n=4, 50%), followed by type IV (n=2, 25%), and type III (n=1, 12.5%). Two patients with type III had the highest mean clinical score (16.0±1.4), whereas type Ia patients had the lowest (7.5±1.8; P=0.003) (Table 1). The mean lumbar BMD-SDS of the mild severity group was -0.7±2.2, which was significantly higher than that of the moderate severity group (-3.7±1.6, P=0.002). Fracture frequency at diagnosis was 0.9±0.6 times per year in the mild severity group and 1.1±1.3 times per year in the moderate severity group (P=0.446). The mean clinical score of 16 patients with blue sclera was 8.6±2.2, which was significantly lower than that of the 8 patients without blue sclera (13.2±2.9, P=0.011). The mean clinical score of 7 patients with DI was 11.6±2.3, which was significantly higher than that of the 17 patients without DI (8.9±3.0, P=0.013). However, there was no significant difference in clinical scores according to presence of hearing loss symptoms (10.8±1.2 vs. 9.4±3.4, P=0.199). The type III patient with the highest clinical score showed skeletal deformity on prenatal ultrasound and was diagnosed with OI at the age of 1 month. Immediately after birth, he was admitted to the neonatal intensive care unit because of respiratory distress. He suffered 50 fractures per year and presented with progressive skeletal deformities, including scoliosis. He was treated with pamidronate for 64 months, starting at the age of 28 months, and at the time of this study, he was still alive at the age of 7.8 years.
2. Molecular analysis of the COL1A1 and COL1A2 genes and genotype-phenotype correlations
Twelve mutations in the COL1A1 and COL1A2 genes were identified in 13 patients from 12 unrelated pedigrees who underwent mutation analysis (Table 2). One mutation was novel, while the other 11 have been previously reported to be pathogenic (7 mutations) or likely pathogenic (4 mutations) [7,23-25]. The novel variant was a heterozygous mutation of c.1984-7_1985del(CCCTCAGGG) at the intron 29-exon 30 boundary of the COL1A1 gene and was classified as quantitative.
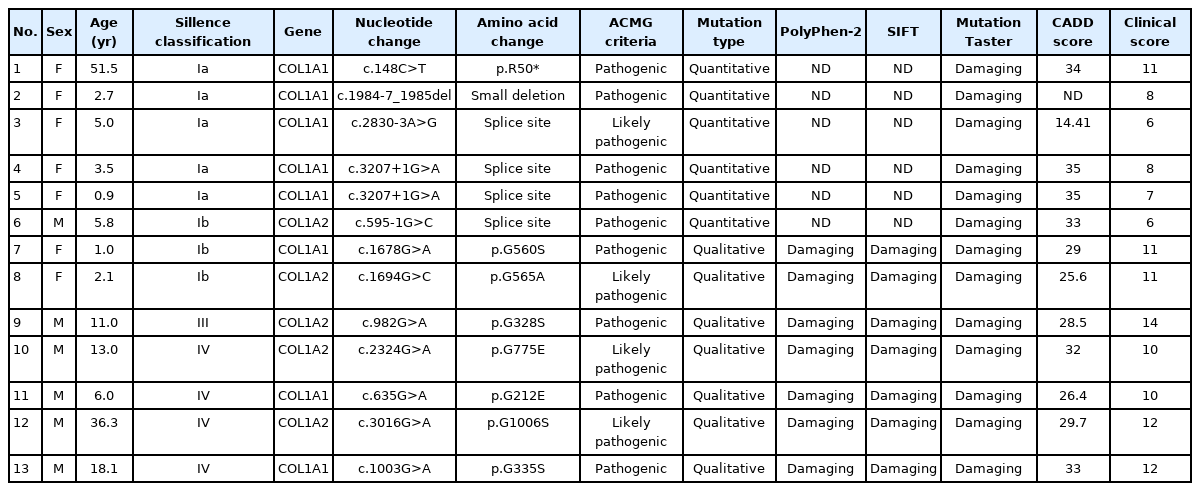
Genetic analysis results of 13 osteogenesis imperfecta patients confirmed by COL1A1 and COL1A2 gene analysis
Five quantitative mutations were identified in 6 patients, and 7 qualitative mutations were found in the remaining 7 patients. Among the quantitative mutations, 1 nonsense mutation (c.148C>T [p.R50*]), 1 small deletion (c.1984-7_1985del), and 3 splice-site mutations were identified. The qualitative mutations were caused by substitution of a Gly within the triplet Gly-X-Y domain of the triple helix. Among these mutations, substitutions with serine were the most common (n=4, 57.1%), followed by glutamic acid (n=2, 28.6%) and alanine (n=1, 14.3%).
The median age at diagnosis was 4.8 years (range, 0.9–51.5 years) in the quantitative mutation group and 13 years (range, 1–36.3 years) in the qualitative mutation group (P=0.445). There were no significant differences between the 2 groups in heightSDS (-1.3±0.9 vs. -2.7±2.4, P=0.295) or weight-SDS (-0.8±0.9 vs. -0.9±1.3, P=0.628) (Table 3).
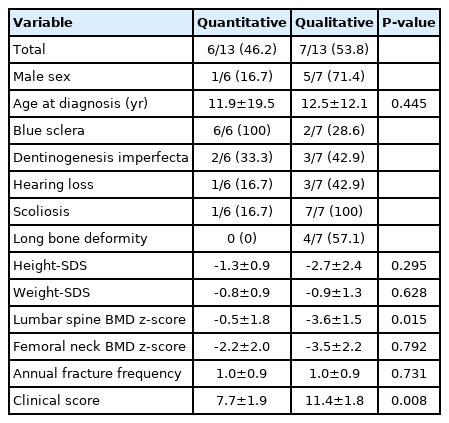
Comparison of phenotypes between quantitative and qualitative mutations in patients with osteogenesis imperfecta
Blue sclera was observed in all patients with quantitative mutations and in 2 of the 7 patients (28.6%) with qualitative mutations (P=0.005). Scoliosis was observed in only 2 of the 6 patients (33.3%) in the quantitative mutation group and in all 7 patients in the qualitative mutation group (P=0.021). Mean lumbar BMD-SDS was significantly higher in patients with quantitative mutations than in those with qualitative mutations (-1.4±4.4 vs. -4.1±2.0, P=0.005). Conversely, there was no statistically significant difference in annual fracture frequency between the 2 groups (1.0±0.9 vs. 1.0±0.9, P=0.731). Mean clinical score was significantly lower in the quantitative mutation group (7.7±1.9) than in the qualitative mutation group (11.4±1.8, P=0.022) (Table 3). The type III patient with the highest clinical score did not undergo genetic analysis. The other type III patient had the second highest score and harbored a c.982G>A (p.G328S) mutation in the exon 19 of the COL1A2 gene.
3. Efficacy and safety of pamidronate treatment in patients with OI
A total of 22 patients received pamidronate treatment: 11 of the 12 (91.7%) type Ia patients, 6 type Ib patients (100%), 2 type III patients (100%), and 3 patients with type IV (75%). Two patients were not treated because they were lost to follow-up. The mean age at the start of treatment was 10.2±9.6 years, and the median age was 6.8 years (range, 1.0–33.2 years). The treatment duration was 30.3±15.7 months (range, 6–64 months). There were no cases of long bone deformity, deterioration, or scoliosis during or after treatment. On the first day of treatment, 4 patients experienced low-grade fever and myalgia, which were relieved with acetaminophen. Mean height-SDS changed from -1.6±1.3 to -1.5±1.3 (P=0.185). Mean fracture frequency decreased from 3.2±10.5 to 0.4±0.8 times per year (P=0.008). Mean BMD-SDS of the lumbar spine improved from -1.6±2.6 to 0.2±2.7 (P=0.001), and that of the femur improved from -3.0±2.3 to -0.9±1.6 (P=0.001; Table 4).

Response to pamidronate treatment in 22 patients with osteogenesis imperfecta
Of the 22 patients who underwent treatment, COL1A1 or COL1A2 mutations were identified in 11: 5 (45.5%) with quantitative mutations and 6 (54.5%) with qualitative mutations. The mean age at diagnosis was 4.0±1.9 years in the quantitative mutation group, and 8.5±6.6 years in the qualitative mutation group (P=0.247). The age at the start of treatment was 5.2±3.1 years (median age, 5.9 years) in the quantitative mutation group and 12.8±10.3 years (median age, 10.7 years) in the qualitative mutation group (P=0.247). Treatment duration was 30.6±18.9 months in patients with quantitative mutations and 27.2±14.2 months in patients with qualitative mutations (P=0.662). Fracture frequency decreased from 1.10±0.91 times per year to 0.0 in patients with qualitative mutations (P=0.027) but did not improve in patients with quantitative mutations. Mean lumbar BMD-SDS increased from -0.5±1.2 to 0.7±2.6 in the quantitative mutation group (P=0.279) and from -2.7±3.0 to -0.6±3.2 in the qualitative mutation group (P=0.028). Mean femur BMD-SDS increased from -2.1±2.1 to -0.5±1.9 in the quantitative mutation group (P=0.068) and from -2.2±0.8 to -1.5±1.7 in the qualitative mutation group (P=0.345). The biochemical parameters of serum calcium, phosphorus, PTH, and osteocalcin levels showed no significant changes before or after treatment in either group (Table 5).
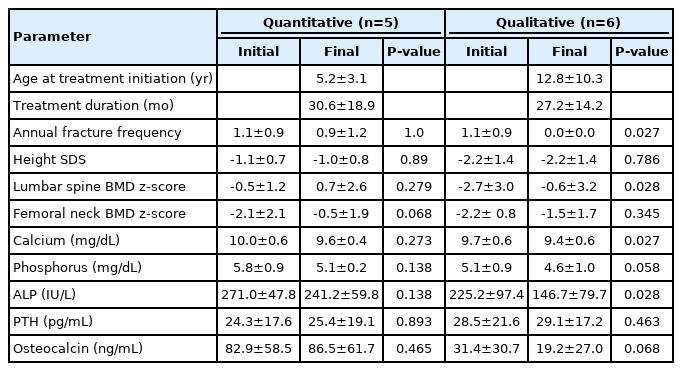
Efficacy of pamidronate treatment according to genotype
The type III patient who reached adulthood was diagnosed with OI at the age of 11 years and was referred to our institute at the age of 30.1 years. After 37 months of pamidronate treatment, his BMD improved significantly. However, his BMD-SDS decreased to -4.0 at the age of 38.3 years. He has been treated with oral alendronate until recently.
Discussion
This study described the clinical phenotypes of 24 Korean OI patients and analyzed the genotype-phenotype correlations in 13 patients using the clinical scoring system. The results showed that long-term intravenous pamidronate treatment was safe and effective in the 22 patients who underwent pamidronate treatment. Type I OI exhibited a milder clinical phenotype and higher BMD-SDS than other types, which is consistent with previous studies [8,23,26-28]. Type Ia patients had the lowest clinical scores, while type III patients had the highest. Interestingly, type Ib patients, who were characterized by DI presence, showed different clinical features than type Ia patients. Mean lumbar BMD-SDS was lower in type Ib than in type Ia patients. The clinical scores of type Ia patients corresponded to mild severity, while those of type Ib patients were significantly higher and corresponded to moderate severity. These differences in clinical severity were correlated with genotypes, i.e., quantitative mutations were associated with mild phenotype and qualitative mutations were accompanied by severe phenotype.
The prevalence of DI in this study was 29.2% (7 of 24), which was similar to previous reports [29,30]. A previous study showed that Gly mutations in 2 collagen type Iα chains, α1(I) and α2(I), are associated with DI, suggesting that DI is a representative phenotype for Gly substitutions [31]. In our study, genetic analysis was performed in 4 of 7 patients with DI, and all of them harbored missense mutations with Gly substitutions. A crosssectional study found that DI occurs mainly in patients with qualitative collagen defects and moderate to severe OI and rarely in patients with quantitative defects and mild OI [32]. In our study, the mean clinical score of 7 patients with DI was significantly higher than that of patients without DI.
OI type I is mainly caused by quantitative mutations, whereas types II–IV are usually caused by qualitative mutations [26,28]. In this study, all 5 quantitative mutations were identified in type I (4 in type Ia and 1 in type Ib). Qualitative mutations were found in 2 type Ib patients, 1 type III patient, and 4 type IV patients. Like in previous studies, the clinical features involving the skeletal system were more severe in the qualitative mutation group [23,26,28]. The mean lumbar spine BMD-SDS of the quantitative mutation group was higher than that of the qualitative mutation group. The mean clinical score of patients with quantitative mutations was significantly lower than that of patients with qualitative mutations, which is consistent with previous studies [26-28,31]. In our study, the clinical scores of 2 type Ib patients with qualitative mutations (p.G560S in COL1A1 and p.G565A in COL1A2) corresponded to moderate severity, while the clinical scores of patients with quantitative mutations corresponded to mild severity. Maioli et al. and Rauch et al. found that clinical severity does not differ between qualitative and quantitative mutations in type I patients [26,31]. This discrepancy may be due to the different mutation spectrum in the study populations, such as α2-serine substitutions [26].
Bisphosphonates, which are analogues of pyrophosphate, attach to hydroxyapatite binding sites on bony surfaces, where active resorption takes place. Bisphosphonates inhibit the bone resorption of osteoclasts and induce their apoptosis by interfering with the mevalonate pathway and synthesis of cholesterol and other isoprenoid lipids. 33) The positive effects of bisphosphonate on spinal deformity and BMD are relatively well known in children with OI. Sato et al. reported a decreased progression of scoliosis in type III patients but not in type I or type IV patients. Moreover, randomized trials have demonstrated that bisphosphonate therapy is effective in increasing BMD in OI [34,35]. Another study reported BMD improvement and decrease in fracture frequency after treatment in type I patients, regardless of genotype [36]. Three previous studies in Korea also found that pamidronate therapy in OI patients increased BMD and reduced the fracture frequency [17,37.38]. In our study, although there was no difference in treatment duration between the 2 mutation groups, lumbar BMD-SDS improved and fracture frequency decreased only in patients with qualitative mutations.
This study has several limitations. Because of its retrospective design and the small number of patients, some clinical data and laboratory findings were not available. During the treatment period, bone pain and quality of life could not be assessed because of lack of data. Moreover, molecular analyses were not performed on all patients, and OI-related genes involved in posttranslational processing/modification (CRTAP, CEPRRE1, PPIB, and TMEM38B), folding, and cross-linking (SERPINH1, FKBP10, and PLOD2) were not analyzed [8]. The International Society of Clinical Densitometry recommends BMD of the spine and total body less head for assessing bone density in children [39]. However, this study utilized BMD of the spine and femur neck because of the lack of data.
In conclusion, OI patients with qualitative mutations in COL1A1 or COL1A2 had lower BMD-SDS and higher clinical scores than those with quantitative mutations, confirming that genotype is helpful for predicting clinical severity of the disorder. The efficacy of pamidronate treatment differed according to genotype: only the qualitative mutation group showed a significant reduction in fracture frequency and an increase in lumbar BMD-SDSs after treatment. Molecular analysis can be useful to predict clinical severity of OI and responsiveness to pamidronate therapy. Because there are genetic heterogeneities and a variable phenotypic spectrum, even within the same mutation, further research is required to verify genotype-phenotype correlations and predictors for clinical severity.
Supplementary Materials
Supplementary Table 1 can be found via https://doi.org/10.6065/apem.2142144.072.
Supplementary Table 1.
Scoring system for patients with osteogenesis imperfecta
Notes
Ethical statement
This study was approved by the Institutional Review Board of Asan Medical Center, Seoul, Korea (No. 2020-1786). Informed consent was waived because of the retrospective nature of the study.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research was supported by a National Research Foundation of Korea (NRF) grant (No. NRF2021R1F1A104593011) and was funded by the Korean Ministry of Science and ICT.
Author contribution
Conceptualization: YC, JHC; Data curation: YC, SH, GHK, BHL, HWY, JHC; Formal analysis: YC, GHK. HWY; Funding acquisition: JHC; Methodology: YC, GHK; Visualization: YC; Writing - original draft: YC, JHC; Writing - review & editing: YC, JHC