The first case of novel variants of the FSHR mutation causing primary amenorrhea in 2 siblings in Korea
Article information

Abstract
Follicle-stimulating hormone receptor (FSHR) mutation is a rare cause of amenorrhea. We report the first case of FSHR mutations in Korea. Two female siblings, aged 16 (patient 1) and 19 (patient 2) years, were referred to the pediatric endocrinology clinic because of primary amenorrhea despite normal breast budding. Gonadotropin-releasing hormone stimulation test showed markedly elevated luteinizing hormone and follicle-stimulating hormone with a relatively low level of estrogen, suggesting hypergonadotropic hypogonadism. Pelvic magnetic resonance imaging revealed a bicornuate uterus in patient 1 and uterine hypoplasia with thinning of the endometrium in patient 2. The progesterone challenge test revealed no withdrawal of bleeding. After two months of administration of combined oral contraceptives, menarche was initiated at regular intervals. To determine the genetic cause of amenorrhea in these patients, whole exome sequencing (WES) was performed, which revealed a compound heterozygous FSHR mutation, c.1364T>G (p.Val455Gly) on exon 10, and c.374T>G (p.Leu125Arg) on exon 4; both of which were novel mutations and were confirmed by Sanger sequencing. The patients maintained regular menstruation and improved bone mineral density while taking combined oral contraceptives, calcium, and vitamin D. Therefore, FSHR mutations can be the cause of amenorrhea in Koreans, and WES facilitates diagnosing the rare cause of amenorrhea.
Highlights
· FSHR mutation should be considered a rare cause of delayed puberty, amenorrhea, and hypergonadotropic hypogonadism in Korea.
· The bicornuate uterus and thinning of the endometrium in our case suggested an association between FSHR mutation and pathology in normal uterine development.
Introduction
Follicle-stimulating hormone (FSH) plays an important role in normal follicular maturation and regulation of estrogen production by granulosa cells in females [1]. The FSH receptor (FSHR) gene (OMIM #136435) is located on chromosome 2p21 and contains 10 exons spanning 54 kb [2]. FSHR mutation can be caused by homozygous or compound heterozygous and activating or inactivating mutations, which are inherited in an autosomal recessive or dominant fashion [1]. To date, 36 mutations in FSHR have been reported in HGMD (HGMD Professional 2020.4 http://www.hgmd.cf.ac.uk/ac/index.php). Among them, missense mutations (80.5%) were the most frequent, followed by regulatory mutations (11.1%). Here, we describe the first Korean cases of FSHR variants manifesting as primary amenorrhea in a pair of siblings. We also provide a literature review of inactivating mutations of FSHR in females.
Case report
Two female siblings aged 16 (patient 1) and 19 (patient 2) years were referred to our pediatric endocrinology clinic because of primary amenorrhea. Secondary sex characteristics were present; Tanner staging of breast/pubic hair was IV/III and III/I for patients 1 and 2, respectively. Breast budding was initiated at the age of 11 years in both patients. The external genitalia was normal in both patients. Anthropometric measurements of patient 1 were within the normal range, but patient 2 had low standard deviation scores for height (-2.31), weight (-2.73), and body mass index (-1.60). Both patients' blood pressure was normal. Abnormal physical examinations, including dysmorphic features, hirsutism, and goiter, were not identified. There was no history of strenuous exercise, abnormal eating behavior, or significant weight changes. One month prior to consultation, patient 1 visited a local clinic because of abdominal pain and underwent abdominal computed tomography, which revealed a bicornuate uterus. Otherwise, there was no specific medical history.
The results of laboratory testing were similar between the siblings. Both patients' hemoglobin levels and thyroid function test results were normal, and their urine human chorionic gonadotropin level was negative. Other hormonal results of testosterone, free testosterone, dehydroepiandrosterone sulfate, prolactin, and sex hormone-binding globulin were within normal limits. The anti-Müllerian hormone level in patient 2 was not low. The FSH level was elevated, and subsequent gonadotropin-releasing hormone stimulation testing showed markedly elevated levels of peak luteinizing hormone (LH) and FSH, suggesting hypergonadotropic hypogonadism in both patients. The adrenocorticotropic hormone stimulation test showed a normal response, which enabled exclusion of congenital adrenal hypoplasia in both patients. The karyotypes of them were 46, XX (Table 1)
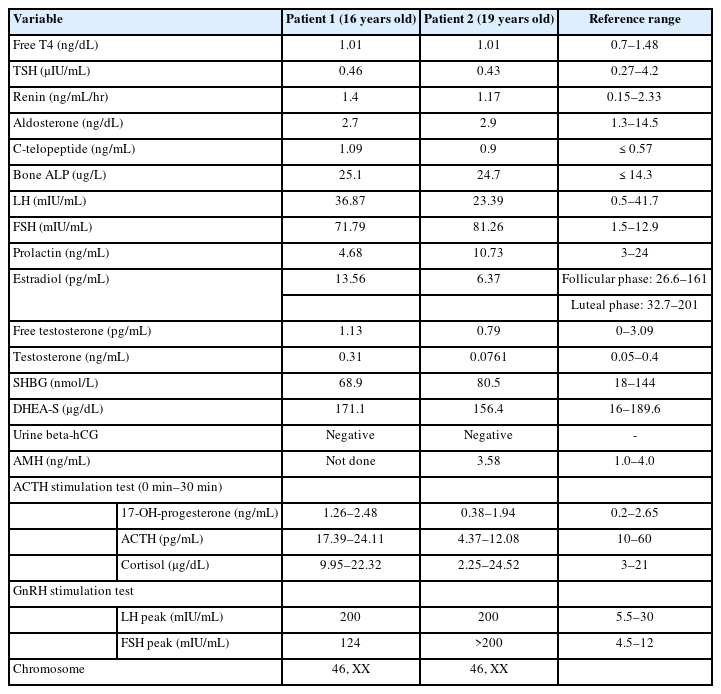
Laboratory and hormonal profile of patient with FSHR mutation
Pelvic magnetic resonance imaging (MRI) showed abnormalities in the reproductive organs of both patients. MRI of patient 1 showed a normal bilateral ovary but a bicornuate uterus with a pair of endometrial canals widely separated by the myometrial septum extending to the internal cervical os. Likewise, MRI of patient 2 confirmed a normally shaped but hypoplastic uterus (1.3 cm×3.7 cm) with endometrial thinning. In addition, ovarian size, shape, and enhancement were normal in both patients. The vaginal tracts were also normal, and there were no abnormalities associated with the urinary tract or kidneys in either sibling (Fig. 1). Bone densitometry showed a low z-score <-2.0 in the femur neck and lumbar spine, compatible with juvenile osteoporosis. The levels of bone turnover markers, including C-telopeptide and bone alkaline phosphatase, were increased (Table 1).

Pelvic magnetic resonance imaging of patients. (A) Sagittal T1-weighted and coronal oblique T2-weighted BLADE sequence imaging of patient 1 revealing a bicornuate uterus (arrow) with a pair of endometrial canals widely separated by the myometrial septum extending to the internal cervical os. Note the fundal cleft of the outer uterine contour. (B) Sagittal T2 BALDE sequence imaging of patient 2 showed hypoplasia of the uterus (1.3 cm×3.7 cm) (arrowheads) with thinning of the endometrium. Size, shape, and enhancement were normal in both ovaries (not seen in this image).
A progesterone challenge test was performed with medroxyprogesterone 5 mg daily for 10 days. However, withdrawal bleeding was not induced, suggesting inadequate production of endogenous estrogen. Combined oral contraceptives (drospirenone and ethynyl estradiol) were administered. Menarche was initiated at regular intervals after 2 months in both patients.
Whole-exome sequencing (WES) was performed to determine the genetic causes of amenorrhea. Genomic DNA was extracted from the patients' buccal swab samples. All exon regions of all human genes (~22,000) were captured using the SureSelect kit (version C2, December 2018; Agilent Technologies, Santa Clara, CA, USA) and were sequenced using the NovaSeq platform (Illumina, San Diego, CA, USA). WES revealed a compound heterozygous FSHR mutation, i.e., c.1364T>G (p.Val455Gly) on exon 10 and c.374T>G (p.Leu125Arg) on exon 4; both of these were novel mutations and were confirmed by Sanger sequencing (Fig. 2A) . Moreover, both siblings maintained regular menstruation and improved bone mineral densities while taking combined oral contraceptives, calcium, and vitamin D.
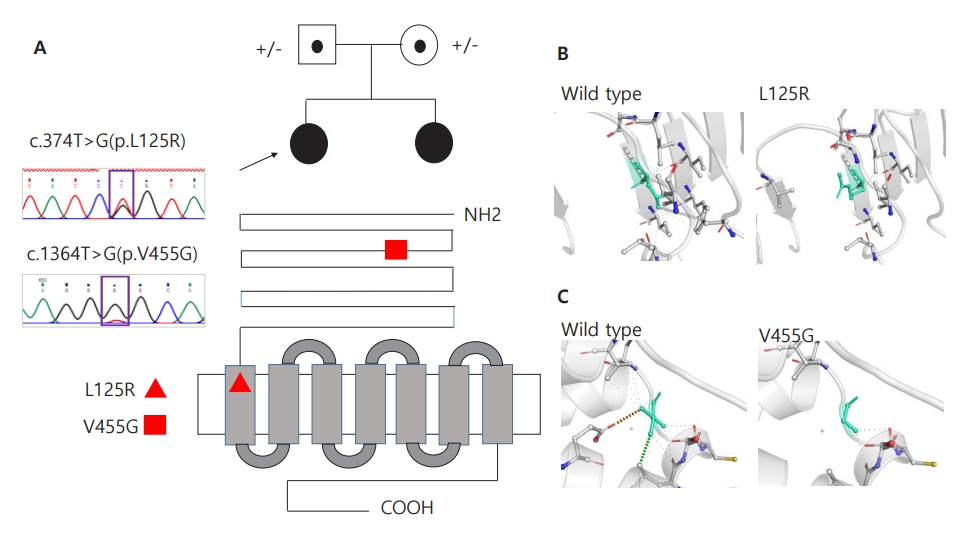
Two novel heterozygous variants in the FSHR gene. (A) Sanger sequencing confirmed novel variants of c.374T>G (p.L125R) or c.1364T>G (p.V455G) in FSHR in 2 siblings, respectively. L125R is located on the leucine-rich repeats domain, and V455G is located on the transmembrane receptor domain, as shown in the red triangle and square. (B, C) Wild-type and mutant residues (p.L125R and p.V455G) in the FSHR protein are shown in light-green and also are represented as sticks alongside the surrounding residues, indicating any type of interaction. Green dots represent hydrophobic contacts with a surrounding residue. Red dots represent hydrogen bonds with a surrounding residue. The crystal structure of the domain from wild-type FSHR was generated by SWISS-MODEL (https://swissmodel.expasy.org/) and has been depicted as a cartoon representation. All structural images were generated using PyMOL (https://pymol.org/).
This study was approved by the Institutional Review Board (IRB) of Pusan National University Yangsan Hospital (IRB No. 05-2021-045). Informed consent was obtained from the patients for publication and genetic analysis.
Discussion
FSH is a dimeric glycoprotein composed of α- and β-subunits released from the anterior pituitary gland [3]. This gonadotropin exerts its gonadal and reproductive function by binding to the FSH receptors of testicular sertoli or ovarian granulosa cells [3]. Although the early stages of follicular growth occur independently, FSH is essential for granulosa cell differentiation, follicular growth, and oocyte maturation [4].
The FSH receptor is a G protein-coupled receptor encoded by the FSHR gene containing 10 exons and 9 introns [2]. Exons 1–9 encode the extracellular domain, while exon 10 encodes the C-terminal part of the extracellular domain, the transmembrane domain, and the intracellular domain [2]. The extracellular domains are responsible for the specificity and high affinity of ligand binding, whereas the transmembrane domains are responsible for G protein receptor activation and signal transduction [3]. In our case, mutations occurred in exons 4 and 10, which are responsible for extracellular membrane and transmembrane domains, respectively. Two of the variants identified in this study (p.L125R and p.V455G in FSHR) have not been previously reported and were absent in the gnomAD and 1000 genome population databases. All variants were predicted to be deleterious by a trio of bioinformatics programs (SIFT: 0.00, 0.02; PolyPhen-2: 1.00, 1.00; and MutationTaster: 0.99, 0.99). Protein crystallization revealed that the p.L125R and p.V455G variants might affect the formation of hydrophobic contacts (Fig. 2B) and that of hydrogen bonds and hydrophobic contacts between the surrounding residues (Fig. 2C), respectively. Moreover, due to structural alterations, the p.V455G variant caused changes in vibration-related entropy change upon mutation, increasing molecule flexibility (ΔΔSVib ENCoM: 0.501 kcal/mol/K), which led to protein destabilization (ΔΔG: -0.465 kcal/mol) based on structure-based predictions (Supplementary Fig. 1).
Fan and Hendrickson [3] demonstrated the nature of FSH-FSH receptor interactions through their crystal structure. Each subunit of FSH binds to the concave face of the curved tube-shaped extracellular domain of the FSH receptor in a direction perpendicular to the hand-clasp fashion, which is accompanied by conformational changes in the α-subunit loops adjusting their shape to reach optimal interaction. FSH-FSH receptor dimerization is followed by various intracellular signaling pathways through activation of the canonical Gs, adenylyl cyclase, cAMP, and protein kinase A [5].
Inactivating FSHR mutations cause altered cell surface expression, decreased binding capacity, and impaired signal transduction and receptor expression. This can lead to delayed puberty, hypergonadotropic hypogonadism, primary or secondary amenorrhea, infertility, ovarian dysgenesis, predisposition to sex cord ovarian tumors in female individuals, and impaired spermatogenesis in male individuals [1].
Existing studies of inactivating FSHR mutations in female individuals are summarized in Table 2, which was created referring to the report by He et al. [6] An inactivating FSHR mutation was first described in Finnish females in 1996 [7]. Most variants were located in the extracellular domain, transmembrane domain, and. extracellular loop of the FSH receptor, while one variant (c.1717C>T) was located in the intracellular loop. In vitro studies have suggested that these variants disrupt cellular functions such as cAMP production, membrane localization, and FSH binding according to variant. The main clinical feature was hypergonadotropic primary amenorrhea. In some cases, Tanner stage III–IV of the underdeveloped breast was noted even in adulthood. Manifestations of the reproductive structure varied. Although there was one case of a normal fertile woman, most showed small-sized ovaries, ovarian dysgenesis, hypoplastic uterus, or endometrial thinning. However, a dysmorphic uterus, such as seen in our case, has not been reported in previous studies. The 5 studies involving histologic biopsy showed follicular arrest up to the small antral stage.

Studies of inactivating mutations of FSHR in female
The bicornuate uterus and thinning of the endometrium in our case suggested an association between FSHR mutation and pathology in normal uterine development. There have been several reports regarding the extragonadal expression of FSHR, suggesting the physiological functions of FSH. Shemesh reported that the gonadotropin receptor showing a dynamic pattern of expression in extragonadal reproductive tissue indicated its substantial role in the molecular autocrine-paracrine regulation of the estrous cycle [8]. Furthermore, LH receptors are expressed in the endometrium, myometrium, and cervix, but FSH receptors are expressed mainly in the cervix [8]. Stilley et al. [9] summarized the distribution of FSH receptors in extraovarian reproductive tissues using various methods. Although there was a discordance in detection method, FSHR protein was observed in the cervix, endometrium, and myometrium. In pregnant women, FSHR protein was observed in the decidua, placental chorionic villi, umbilical cord, amnion, vascular smooth muscle, and endothelium [9]. Although these studies support the role of FSH in regulating uterine contractile activity, implantation, and the estrous cycle, they do not demonstrate the role of FSH and its receptors in uterine development. One experimental study conducted on FSHR-haploinsufficient aged mice reported uterine abnormalities, such as a nodular structure, cyst, hypertrophic epithelium, and increased angiogenesis. The authors concluded that an imbalance in progesterone receptor isoforms A and B and increased LH receptors in the uterus may have contributed to a high frequency of uterine pathology [10]. Increased gonadotropin in fetuses with FSHR mutation due to gonadal failure and a wide distribution of gonadotropin receptors in reproductive tissues may lead to uterine dysmorphism. However, further research is needed to identify the pathophysiology of the uterus in FSHR mutations.
As with osteoporosis in the 2 siblings in the present case, hypogonadism is highly associated with bone loss. Interestingly, Sun et al. [11] showed that neither FSH-β nor FSH receptor null mice exhibited bone loss despite severe hypogonadism. In contrast, increased bone mass and decreased osteoclastic resorption were obser ved in haploinsufficient FSHβ+/− mice with normal ovarian function. They concluded that the skeletal action of FSH was estrogen-independent, and high circulating FSH level can cause hypogonadal bone loss. Based on these results, osteoporosis in individuals with FSHR mutation is supposed to be more severe than that in those with hypogonadism without FSH or FSHR pathology. However, further research is required.
Several previous studies conducted in Korea have demonstrated rare prevalence of FHSR mutations. A Korean study comparing infertile and healthy fertile women showed variant types of FSH receptor exon 10 (c.919A>G), but the authors concluded that this mutation may not be specific to premature ovarian failure patients [12]. Another Korean study determining the presence of an FSHR mutation (c.566C>T) could not identify any mutation in the FSHR gene in either the premature ovarian failure group or the healthy group [13,14]. Therefore, our study is the first report of FSHR mutation causing primary amenorrhea in Korea.
According to our case report, FSHR mutation should be considered a rare cause of amenorrhea in Koreans, and WES gave us the chance to diagnose a rare cause of amenorrhea. Individuals with FSHR mutations can manifest ovarian and uterine pathologies requiring proper radiologic investigation. In addition, they can be more susceptible to osteoporosis than other patients with hypogonadism; therefore, careful management is needed.
Supplementary Materials
Supplementary Fig. 1 can be found at https://doi.org/10.6065/apem.2142116.058.
Supplementary Fig. 1.
Visual representation of the Δ entropy energy between the wild-type and p.V455G variant in which the amino acids were colored according to the vibrational entropy change upon mutation. Results from other predictive tools (NMA-based and other structure-based approaches) are also displayed to predict the mutation effect using the Dynamut web server with the normal mode analysis function (http://biosig.unimelb.edu.au/dynamut/). Red represents the gain in flexibility.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: CKC; Data curation: SY, JYY; Formal analysis: SY, CK, CKC; Methodology: CKC; Visualization: CK; Writing - original draft: SY; Writing - review & editing: JYY, CKC