Prader-Willi syndrome: an update on obesity and endocrine problems
Article information

Abstract
Prader-Willi syndrome (PWS) is a rare complex genetic disorder that results from a lack of expression of the paternally inherited chromosome 15q11-q13. PWS is characterized by hypotonia and feeding difficulty in early infancy and development of morbid obesity aggravated by uncontrolled hyperphagia after childhood and adolescent. Dysmorphic facial features, delayed motor and language development, various degrees of cognitive impairment, and behavioral problems are common in PWS. Without early, intensive nutritional therapy along with behavioral modification, PWS patients develop severe obesity associated with type 2 diabetes, obstructive sleep apnea, right-side heart failure, and other obesity-related metabolic complications. Hypothalamic dysfunction in PWS can lead to several endocrine disorders, including short stature with growth hormone deficiency, hypothyroidism, central adrenal insufficiency, and hypogonadism. In this review, we discuss the natural history of PWS and the mechanisms of hyperphagia and obesity. We also provide an update on obesity treatments and recommendations for screening and monitoring of various endocrine problems that can occur in PWS.
Highlights
Understanding the mechanisms that lead to hyperphagia and early nutritional intervention with behavioral modifications are important for PWS management. Pediatric endocrinologists should be aware of the recommendations for endocrine problems that can occur in PWS.
Introduction
Prader-Willi syndrome (PWS, OMIM #176270) is a complex genetic disorder caused by lack of expression of the paternally inherited chromosome 15q11-q13 [1]. There are three major molecular classes in PWS: paternal deletion of the chromosome 15q11-q13 region (the most common type of PWS, 65%–75%), maternal uniparental disomy of chromosome 15 (20%–30%), and imprinting defects caused by epimutations or microdeletions in the imprinting center of the chromosome 15q11-q13 region (1%–3%) [2-4]. In addition to these major classes, there are very rare balanced translocations involving the PWS region on chromosome 15q11-q13 (0.1%) [5]. Previous epidemiologic studies have estimated the incidence of PWS from 1 in 10,000 to 1 in 30,000 live births [6-8]. Clinically, PWS is characterized by hypotonia and feeding difficulty in early infancy, and development of hyperphagia exacerbated by impaired satiety from late infancy or childhood is characteristic of PWS.
Without aggressive control of excessive eating behaviors, PWS patients develop severe obesity associated with type 2 diabetes mellitus (T2DM), obstructive sleep apnea, right-side heart failure, and other obesity-related metabolic complications later in life. Dysmorphic facial features, delayed development of motor and language skills, various degrees of cognitive impairment, and behavioral problems are common in patients with PWS. Hypothalamic dysfunction can lead to hyperphagia, obesity, and several endocrine disorders, including short stature with growth hormone deficiency (GHD), hypothyroidism, central adrenal insufficiency (CAI), and hypogonadism in PWS patients [9,10]. This review aims to provide an update of the obesity caused by hyperphagia and the endocrine problems present in PWS.
Natural history of PWS
The natural course of PWS has several nutritional phases with gradual and complex progression [11]. Fig. 1 summarizes the nutritional stages of PWS. During Phase 0, prenatal features of PWS include decreased fetal activity, polyhydramnios, breech presentation, small for gestational age, and lower birth weight than siblings [12]. In phase 1 (at birth), hypotonia is noted. During phase 1a (0–9 months), poor sucking and feeding difficulties with/without failure to thrive develop. In phase 1b (9–24 months), the infant grows steadily along a disease-specific growth chart [13,14]. Phase 2 involves the transition from anorexia to excessive weight gain. In phase 2a (2–4.5 years), weight gain without a significant change in appetite occurs. During phase 2b (4.5–8 years), appetite tends to increase along with interest in food. Phase 3 (8 years–adult) onward is marked by hyperphagia with uncontrolled appetite accompanied by a lack of satiety and food-seeking behavior.
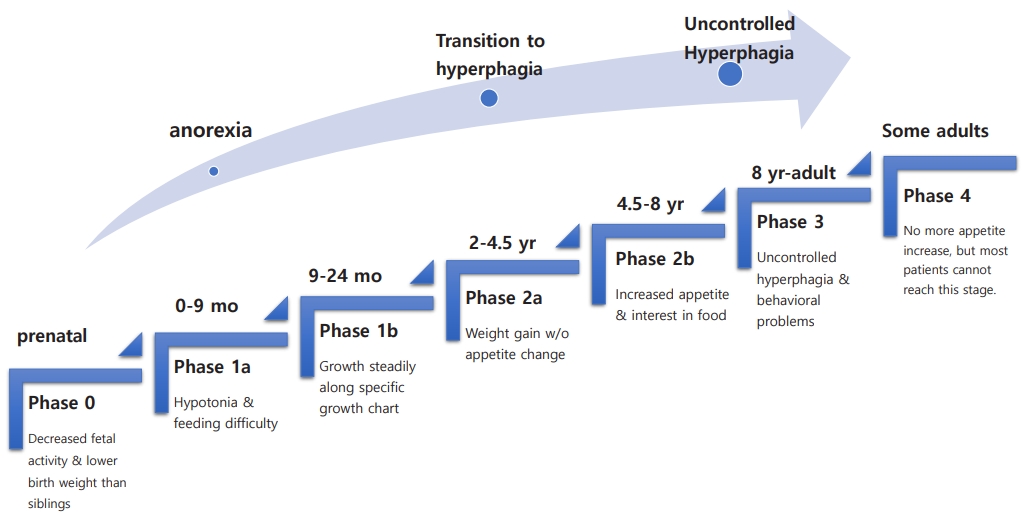
The nutritional stages of Prader-Willi syndrome.
Some but not most adults move on to phase 4, where they no longer have an increased appetite. The precise mechanism by which the nutritional phases progress is unknown. As these stages progress, various behavioral and endocrine problems arise. Comorbidities accompany these patients throughout their lives. Management of PWS requires a team approach, including a neonatologist, medical geneticist, pediatric endocrinologist, endocrinologist/diabetologist, psychologist, orthopedist, specialized dietitian, speech therapist, physical therapist, psychotherapist, social worker, etc [1,15-17].
Despite recent improvements in multidisciplinary care of patients with PWS, its morbidity and mortality rates increase with age [18,19]. According to previous observational studies, the mean age of death in patients with PWS was 29.5±16 years (range, 1 month–67 years), and respiratory problems (including respiratory failure and pulmonary infection) were the most common causes of death at all ages. The other causes of death in PWS were cardiovascular disease, gastrointestinal causes, infection, choking events, and accidents [19-21].
Hyperphagia and obesity in PWS
1. Mechanism of hyperphagia and obesity in PWS
Understanding the pathophysiology of hyperphagia and obesity in PWS is very important for the care of patients with PWS. Although research has investigated the mechanisms of hyperphagia and obesity in PWS, much remains unclear. Hypothalamic dysfunction might affect appetite control, energy expenditure, and orexigenic/anorexigenic hormone regulation, leading to hyperphagia and obesity in PWS [22-24].
Studies using functional magnetic resonance imaging (fMRI) data showed hyperactivation in response to appealing food or hypoactivation in response to disliked food in the hypothalamus of PWS patients compared with controls [25-27]. These findings suggest that dysfunction in the subcortical reward circuitry and the cortical inhibitory regions associated with appetite and behavioral control in the hypothalamus is involved in development and progression of hyperphagia and obesity in PWS.
Ghrelin is a gut hormone secreted by the gastric mucosa. Acylated ghrelin (AG) is a representative orexigenic hormone that is activated by binding to the growth hormone secretagogue receptor 1a (GHSR1a). Unacylated ghrelin (UAG) is present in the circulation and has the opposite effect of AG. In PWS, high serum level of total ghrelin (especially AG) was persistent even after food intake, which can cause a delay in the sense of fullness and lead to hyperphagia and obesity. Conversely, high UAG level was observed in PWS infants, which might be associated with anorexia in the early nutritional phase (phase 1) before the onset of hyperphagia [28-31].
Oxytocin, a neuropeptide produced in the paraventricular nucleus (PVN) of the hypothalamus, is involved in the homeostatic control of satiety, energy balance, social interaction, and obsessive-compulsive behaviors as well as uterine contractions and lactation promotion [32]. Previous studies have revealed a reduced volume of PVN in the hypothalamus and higher serum and cerebrospinal fluid levels of oxytocin in PWS patients compared with controls [33-35]. Recently, it has been suggested that the GHSR1a receptor and the oxytocin receptor heterocomplex interact to alter the signaling pathways that regulate appetite and satiety [36,37]. Several clinical trials that use these analogs to control appetite in PWS patients are being conducted, but no consistent conclusion has been achieved [38-40]. The roles of ghrelin and oxytocin in appetite regulation and obesity in PWS remain unclear.
Other studies that have examined the roles of several anorexigenic or adipose-derived hormones related to the pathophysiology of obesity have been reported with mixed results in PWS patients (Table 1) [41-49]. The factors that regulate appetite and obesity are complex, and substances other than hormones influence the interactions of these various factors. Therefore, further studies are needed.

anorexigenic or adipose-derives hormones in Prader-Willi syndrome (PWS)
Recent studies using genome-wide transcriptomic analysis of the PWS hypothalamus are emerging to explain the mechanisms behind hyperphagia and obesity in PWS [50]. Bochukova et al. suggested that the hypothalamus in PWS patients showed widespread transcriptomic changes in gene expression and alternative splicing. They indicated that brain-derived neurotrophic factor deficiency has a potential role in developmental delay, hyperphagia, and obesity in PWS [51-53].
Compared to simple obesity, PWS patients have less lean body mass and more fat mass, mainly distributed in the trunk, and less visceral obesity [54,55]. These findings indicate that reduced resting energy expenditure (REE) might promote obesity in PWS patients [22]. Some studies have hypothesized that altered gene expression in the mitochondria play a role in the reduction in energy metabolism observed in PWS [56,57].
Considered together, hyperphagia and obesity in PWS are thought to be caused by complex mechanisms of hypothalamic dysfunction, hormonal circuits involved in appetite or satiety control, changes in body composition, and decreased REE.
2. Management of hyperphagia and obesity in PWS
Obesity control is the most important goal in PWS treatment. However, this task is difficult due to patient decreased lean body mass, reduced REE, and behavioral problems related to appetite control. Early dietary intervention and nutritional counseling are critical in preventing excessive weight gain and development of morbid obesity. The first months of life require dietary treatment for adequate nutrition to prevent malnutrition and failure to thrive. Exercise, behavioral modifications, and a balanced, low-calorie diet can prevent obesity and its complications. Schmidt et al. [58] reported that early dietary treatment with strict fat reduction beginning at the age of 2 years was effective in preventing excessive weight gain after 10 years of age. Infants and children with PWS should consume 60%–80% of the recommended daily allowance to maintain their body weight due to a reduced REE [15,17,59]. Because some children with PWS also exhibit food-seeking and food-stealing behaviors, strategies such as environmental barriers to food access (locking the kitchen, refrigerator, or cupboards) and restricting access to money and food are needed [60].
To date, pharmacologic treatment of hyperphagia and obesity in PWS has been limited. From the 1990s to the early 2010s, drugs of various mechanisms have been tested to control appetite and obesity in PWS patients, including beta endorphin antagonists (such as naltrexone), pancreatic lipase inhibitors (such as orlistat), insulin sensitizers (such as metformin), nonspecific inhibitors of serotonin and norepinephrine (including sibutramine), Na+ channel/gamma-aminobutyric acid modulators (topiramate), and endocannabinoid 1 receptor antagonists (rimonabant, etc.) [60,61]. However, no drugs have been proven effective, and some have been withdrawn from the market due to side effects. Early initiation and long-term use of growth hormone (GH) treatment increases muscle mass and reduces body mass index (BMI) in these patients but is ineffective at controlling appetite or food-seeking behavior. Furthermore, most PWS patients who have been treated with GH remain obese [62].
Bariatric surgery is rarely performed in PWS patients because they are at high risk for gastric necrosis or rupture due to delayed gastric emptying and a decreased sensation of fullness [60,63].
3. Clinical trials of hyperphagia and obesity in PWS
Several ongoing or completed randomized controlled trials (RCTs) for treatment of hyperphagia and obesity in PWS have included a glucagon-like peptide-1 agonist, a melanocortin 4 receptor (MC4R) agonist, diazoxide, oxytocin/carbetocin, UAG analog (AZP-531), etc.
Some case reports and clinical trials have reported that treatment with a GLP-1 agonist, such as exenatide or liraglutide, had beneficial effects on obesity, satiety, and glycemic control in PWS patients [64-66]. Fintini et al. [67] reported that 24 months of subcutaneous liraglutide treatment showed a tendency to decrease BMI, glycosylated hemoglobin (HbA1c), and waist circumference in six obese PWS adults with T2DM, and it was well-tolerated. Salehi et al. [64] found that six months of exenatide treatment decreased the appetite scores and HbA1c level in 10 obese PWS adolescents, but weight, BMI, adiposity, and ghrelin did not change. Large-scale, randomized-controlled, long-term studies on the effects and safety of GLP-1 agonists are needed. No GLP-1 agonist has been approved for children and adolescents in Korea.
Setmelanotide, an MC4R agonist also known as RM-493, is a synthetic peptide that binds strongly to human MC4R. Its side effects include headache, arthralgia, or skin darkening. It has no effects on heart rate or blood pressure and has been shown to increase REE and fat oxidation in obese individuals. However, a phase 2a trial in obese PWS patients (ClinicalTrials.gov: NCT02311673) showed only modest changes in hyperphagia without any reduction in body weight [60].
Diazoxide is a potent K+-ATP channel agonist with a well-characterized safety profile. Diazoxide choline controlled-release (DCCR) is hypothesized to reduce the synthesis and secretion of appetite stimulatory neuropeptides in the arcuate neuron, thereby reducing hyperphagia in PWS. In a phase 2 study, DCCR reduced appetite and aggressive behaviors, improved insulin sensitivity, and decreased waist circumference and fat mass in PWS adults [68]. A multicenter phase 3 clinical trial (ClinicalTrials.gov: NCT03440814) with DCCR for PWS is underway.
As previously mentioned, abnormalities in the oxytocin system are thought to play some role in appetite control and behavioral problems in PWS [32-35]. Several RCTs using oxytocin or its analog (intranasal carbetocin) have been conducted in PWS patients. No evidence suggests that intranasal oxytocin improves symptoms and hyperphagia in PWS [39,69,70]. However, improvements in oral feeding skills have been observed in very young PWS infants who received a short course of oxytocin within the first 6 months of life [71]. A recently completed RCT (ClinicalTrials.gov: NCT01968187) reported intranasal carbetocin was well-tolerated and improved hyperphagia and behavioral symptoms in PWS patients [72].
Ghrelin exhibits unique concentration changes in PWS and might be a pharmacotherapeutic target for these patients. In a phase 2 RCT, a daily subcutaneous injection of a UAG analogue (AZP-531) improved the appetite score, waist circumference, and fat mass in 47 PWS patients [40]. In addition, a phase 2 RCT (ClinicalTrials.gov: NCT03274856) using GLWL-01, a ghrelin O-acyltransferase inhibitor that blocks the conversion of ghrelin to AG, was recently completed in PWS patients, and the results are awaiting analysis. Drugs that affect the ghrelin system could be new treatment options for ameliorating hyperphagia in PWS patients if the effect is consistently demonstrated in large-scale, long-term follow-up studies.
Although no drug yet has shown consistent efficacy in hyperphagia and obesity in PWS, the results of completed or ongoing RCTs deserve attention.
Endocrine problems in PWS
1. Growth hormone deficiency
GHD is the most commonly reported endocrine problem that occurs in 40%–100% of PWS patients. The exact mechanism of GHD is unclear. PWS is associated with reduced 24-hour GH secretion, insufficient response to GH stimulation tests, and a decreased serum IGF-1 concentration [23,73,74]. Children diagnosed with PWS should receive GH treatment (GHT) without GH stimulation tests by as early as 3 to 6 months of age [73,75]. GHT in children with PWS increases their final height and compensates for metabolic disorders caused by GHD. Some studies have reported that early GHT improves motor and cognitive development, socialization, and body composition [75-77]. In Korea, GHT is currently covered by the national health insurance for children over 2 years of age who have been diagnosed with PWS. Given the recent trend toward PWS diagnoses in younger children, allowing infants to receive GHT should be considered.
GHT has growth and metabolic benefits and is relatively safe and well-tolerated in children and adolescents with PWS. Therefore, this population should receive GHT until the end of their linear growth period. Based on the benefits of GHT in PWS, some suggest that it be continued in adulthood or offered to previously untreated adults with PWS [78].
Recent studies have demonstrated that GHT cessation led to a deteriorated physical and social status and increases in visceral fat, low-density lipoprotein cholesterol, and total cholesterol levels in young adult PWS patients [79,80]. In 2020, Damen et al. [81] reported that continued GHT in young adults with PWS maintains the positive effects on body composition attained during childhood. However, since not all adults with PWS meet the diagnostic criteria for GHD by GH stimulation tests [82], an appropriate GH stimulation test method must be established. Further studies are needed to clarify the indications, benefits, and adverse effects of GHT in patients of a transitional age or adults with PWS.
2. Hypothyroidism
Hypothalamic dysfunction can be associated with central hypothyroidism; the prevalence of hypothyroidism in PWS patients ranges from 2%–4% to 20%–30% [83-85]. Vaiani et al. [86] reported that 72% of PWS infants showed low free T4 level and transient or definite thyroid axis dysfunction during their first 2 years of life. Several clinical guidelines recommend that thyroid function be evaluated at the time of the PWS diagnosis and then again annually, especially during GHT. These cases should be treated with levothyroxine immediately if hypothyroidism is present [15,17,85].
3. Central adrenal insufficiency
The prevalence of CAI in PWS patients has been reported to be 0%–60% [87-90]. Farholt et al. [89] revealed a normal cortisol response to high-dose adrenocorticotropic hormone (ACTH) and insulin-tolerance testing (ITT) in 65 children and adults with PWS. Corrias et al. [90] showed that 14.3% (12 of 84) of PWS children had a pathological cortisol peak response to the low-dose ACTH stimulation test. de Lind van Wijngaarden et al. [87] found that 60% of patients with PWS demonstrated a low peak ACTH response on the metyrapone test and met the diagnostic criteria for CAI. In addition, Oto et al. [91] reported that a peak response of cortisol to ITT was delayed in 64% (23 of 36) of PWS patients, which complicated diagnosis of CAI in PWS. Although clinically distinct CAI patients are rare in PWS, the possibility of latent adrenal insufficiency cannot be excluded, as PWS patients with hypothalamic dysfunction are at risk for CAI. Muscogiuri et al. [92] recommended an evaluation of adrenal function before major surgery or anesthesia and use of glucocorticoids if severe hypotension develops during surgery in patients whose normal adrenal function is unconfirmed. However, in patients with CAI unconfirmed by hormone tests and clinical manifestations, chronic hydrocortisone supplementation is not necessary given its negative effects on bone mineral density, obesity, and glucose metabolism [10,92,93].
4. Hypogonadism
Hypogonadism is a typical clinical fe ature of PWS patients regardless of sex and age and is a combination of hypogonadotropic hypogonadism and primary gonadal failure. PWS patients normally go through a mini puberty. Although there are individual differences in the onset of puberty, this event occurs in a relatively typical age range [10,92]. With the onset of puberty, the concentrations of sex hormones (testosterone and estradiol) increase but remain lower than the normal ranges. The serum levels of luteinizing hormone, follicle stimulating hormone, and inhibin B can vary due to the mixed nature of hypogonadism. Therefore, the clinical presentation of hypogonadism in PWS patients is highly variable.
In male PWS patients, cryptorchidism (unilateral or bilateral) is very common and is reported in 66%–100% of newborns; most require orchiopexy [92,94]. Male PWS patients often have thick suprapubic fat pads and a smaller penis size than healthy individuals. This condition is more severe in obese patients and sometimes makes it difficult to urinate while standing [10]. Most male PWS patients begin puberty at a normal age range, but early or delayed puberty has also been reported. However, the progression of puberty usually arrests at Tanner stage III, and testicular size is smaller than that of a normal adult male. Men with PWS are thought to be infertile, and no cases of a male with PWS fathering children have been reported [93]. To date, hypogonadism management guidelines for PWS patients have not been established. Negative effects on skeletal maturation and behavioral problems must be considered when attempting testosterone replacement treatment for a male with PWS [3,10,15,17,92].
Hypoplasia of the labia majora and clitoris at birth is noted in 76% of females with PWS [94]. Puberty in females with PWS begins at a normal age but is delayed in Tanner stages III–IV; 8%–25% of these patients experience spontaneous periods. The average age at menarche is delayed to 20 years [95]. Although most females with PWS have subsequent oligomenorrhea or secondary amenorrhea after menarche, they can become reproductive. Six pregnancies have been reported in women with PWS [10]. Angelman syndrome can occur if a PWS patient with a 15q11.2-q13 deletion gives birth to a child, so education and genetic counseling is required for all female patients with PWS [96]. Although there is no consensus on the utility of hormone replacement therapy in this population, treatment should consider the potentially positive effects on bone density and body image and also the negative effects on skeletal maturation [3,10,15,17].
The prevalence of premature adrenarche has been reported in up to 30% of PWS patients. Premature adrenarche is associated with pubic/axillary hair, advanced bone age, and increased serum dehydroepiandrosterone sulfate level. This condition is generally considered benign, and further tests or treatment are typically not required [9,92,93].
Conclusion
PWS is a complex genetic disorder characterized by uncontrolled appetite, severe obesity, and multiple endocrine problems. Understanding the mechanisms that lead to hyperphagia and obesity in PWS is critical to understanding and managing patients with PWS. So far, early intensive nutritional therapy with behavioral modifications is the best method to control appetite and obesity in PWS. However, several clinical trials investigating appetite suppression and obesity control are ongoing. We hope to attempt pharmacotherapy in the near future to address these issues. Proper management of PWS patients requires a multidisciplinary team approach. It is important for pediatric endocrinologists to be aware of the recommendations for screening and monitoring of various endocrine problems that can occur in PWS (Table 2).
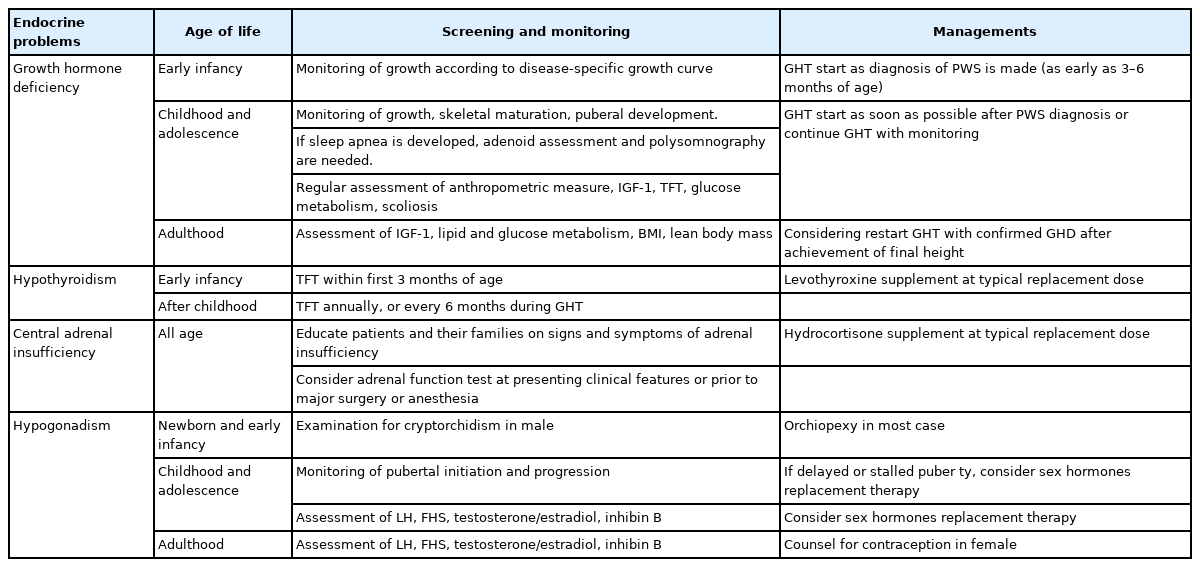
Recommendations for endocrinologic problems in Prader-Willi syndrome (PWS)
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.