Identification of a novel point mutation in DAX-1 gene in a patient with adrenal hypoplasia congenita
Article information

Abstract
X-linked adrenal hypoplasia congenita caused by a mutation in NR0B1/DAX-1 is a rare inherited disorder. Patients with adrenal hypoplasia congenita are usually diagnosed with primary adrenal insufficiency in infancy or early childhood and present hypogonadotropic hypogonadism during adolescence. Our patient first presented with adrenal crisis at the age of 2 months, which was managed with glucocorticoids and mineralocorticoids. At the age of 17 years, testicular volumes of 5 mL each and a stretched penile length of 4 cm were noted. A combined pituitary function test showed a peak luteinizing hormone level of 2.68 mIU/mL, testosterone 13.5 ng/dL, confirming hypogonadotropic hypogonadism. After whole-exome sequencing, a new variant of DAX-1, c.881T>C (p.Leu294Pro), was found. He was diagnosed with X-linked adrenal hypoplasia congenita and then treated with human choriogonadotropin for the induction of spermatogenesis as well as with steroid replacement therapy.
Highlights
A new variant of DAX-1, c.881T>C (p.Leu294Pro) causing X-linked adrenal hypoplasia congenita was found. In this case, the disease etiology was unclear until a genetic analysis was performed. A correct and early diagnosis is important for long-term treatment.
Introduction
X-linked adrenal hypoplasia congenita (OMIM: 300200) caused by gene mutations in DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) is a rare inherited disorder of the adrenal gland [1,2]. The DAX-1 gene, also known as the NR0B1 gene, is located on the short arm of the X chromosome and encodes an orphan nuclear hormone receptor. DAX-1 is expressed in the hypothalamus, pituitary gland, adrenal gland, and gonads. Accordingly, mutations in this gene result in adrenal insufficiency and hypogonadotropic hypogonadism and typically occur in males. Impaired spermatogenesis due to this mutation has also been observed. However, the severity and clinical phenotype can differ between patients.
Here, we report a case of adrenal hypoplasia congenita in a 17-year-old Korean boy with a novel mutation in the NR0B1/DAX-1 gene. Our patient's main clinical manifestations were adrenal insufficiency and hypogonadotropic hypogonadism.
Case report
A 2-month-old male presented with recurrent vomiting and poor oral intake with irritability. He was born via cesarean section at 41-week gestation and weighed 3.9 kg. There were no complications during the perinatal period. The patient had no family history of chronic illness. On physical examination, the patient was dehydrated and showed hyperpigmentation throughout his body. This pigmentation first appeared 2 weeks after birth. The patient's blood pressure was 86/46 mmHg (<50th percentile), with a heart rate of 134 beats per minute and a respiratory rate of 30 breaths per minute. He was 41 cm tall (50th–75th percentile) with a weight of 6 kg (25th–50th percentile) and a body mass index of 14.43 kg/m2 (5th–10th percentile). Laboratory tests revealed hyponatremia (113 mmol/L), hyperkalemia (8.0 mmol/L), elevated adrenocorticotropic hormone (ACTH, 7,207.3 pg/mL) and a relatively low level of cortisol (5.4 μg/dL), indicating primary adrenal insufficiency. Subsequently, the replacement of hydrocortisone and fludrocortisone was commenced. Other initial results before treatment were as follows: 17-hydroxyprogesterone (17-OHP), 0.44 ng/mL; serum aldosterone level, 14.2 ng/dL; and plasma renin activity, 0.26 ng/mL/hr. Other basal hormone levels were within normal ranges (Table 1). Additional studies were performed to evaluate the etiology of the patient's adrenal insufficiency. The result of the adrenal cortex antibody test was negative, and the result of a very long-chain fatty acid test for adrenoleukodystrophy was not remarkable. However, the adrenal glands were not delineated as observed using pelvic ultrasound images. The patient was transferred to another hospital to be closer to his place of residence.

Initial laboratory study findings at the age of 2 months
When he was 17 years old, the patient visited our hospital again for evaluation of his small testes and penis. At that time, his height was 163.5 cm (5th–10th percentile), and his body weight was 70 kg (50th–75th percentile). On physical examination, both testes were 5 mL in volume, and his stretched penile length was 4 cm, indicating androgen deficiency. A chromosome study showed a normal male karyotype with 46, XY. In a human choriogonadotropin stimulation test, the stimulated testosterone to dihydrotestosterone ratio was 7.24, and the basal and stimulated testosterone levels were 13.5 ng/dL and 239.2 ng/dL, respectively (Table 2). In an ACTH stimulation test, the peak cortisol level was below 0.4 µg/dL, indicating cortisol deficiency. The levels of 17-OHP and 17-hydroxypregenenolone were also lower than normal, a result that is not compatible with congenital adrenal hyperplasia due to 21-hydroxylase deficiency or 3β-hydroxysteroid dehydrogenase deficiency (Table 3). Genetic analysis did not identify any mutations in the StAR gene. In a combined pituitary function test, the peak luteinizing hormone level was 2.68 mIU/mL, and the testosterone level was 13.5 ng/dL, consistent with hypogonadotropic hypogonadism. The cortisol level was uncheckable, and the peak growth hormone level was 7.09 ng/mL (Table 4).

Patient's human choriogonadotropin stimulation test results at the age of 17 years

Patient's adrenocorticotropic hormone stimulation test results at the age of 17 years
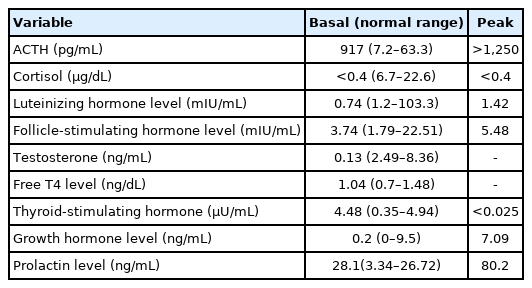
Patient's combined anterior pituitary function test results at the age of 17 years
Genetic analysis for identifying the cause of adrenal insufficiency and hypogonadotropic hypogonadism using whole-exome sequencing showed a mutation in the DAX-1 gene. Whole-exome sequencing was performed using the TruSight One panel of 4,813 genes (Illumina, San Diego, CA, USA) to screen for pathological mutations. The newly discovered variant was c.881T>C (p.Leu294Pro), and this missense mutation was predicted to be deleterious by sorting intolerant from tolerant (SIFT) [3] and as probably damaging by PolyPhen-2 [4]. The variant was not found in ExAC (http://exac.broadinstitute.org/), Clin Var (http://www.ncbi.nlm.gov/clinvar/), nor dbSNP (http://www.ncbi.nlm.nih.gov/project/SNP). Genetic analysis was not performed for the patient's family because they refused to give a blood sample. The diagnosis was confirmed as adrenal hypoplasia congenita. To induce spermatogenesis, exogenous human choriogonadotropin together with follicle-stimulating hormone were prescribed, as well as the existing hydrocortisone and fludrocortisone replacement.
Discussion
X-linked adrenal hypoplasia congenita is a rare inherited disease caused by mutations in the NR0B1/DAX-1 gene. The estimated incidence of NR0B1-related X-linked adrenal hypoplasia congenita in the general population is very low (1:70,000-1:1,2000,000) [5,6]. No specific populations are known to be at risk for this disorder. DAX-1 is expressed throughout the hypothalamo-pituitary-gonadal and adrenal axes and plays an important role in the development of these endocrine organs [7]. Animal studies using DAX-1 knockout mice have shown progressive adrenal insufficiency to be a consequence of the loss of its function. Li et al. [8] demonstrated a reduction in gonadotropin-releasing hormone expression in DAX-1 mutants using a luciferase assay. In addition, DAX-1 knockout mice showed altered testicular architecture and impaired spermatogenesis [9]. However, normal function of the hypothalamic-gonadotrope-Leydig cell axis during the minipuberty of infancy with this disease was reported; therefore, abnormalities in this axis may develop later in life [10,11]. In adrenal hypoplasia congenita, the production of pituitary hormones other than gonadotropins is intact, but a low peak level of growth hormone was observed in our patient. This could be explained by inadequate sex hormone priming before the combined pituitary function test. Most patients with X-linked adrenal hypoplasia congenita present with symptoms of adrenal insufficiency in infancy or early childhood, including skin pigmentation, hypoglycemia, salt wasting dehydration, and failure to thrive [5]. In our patient, the clinical manifestations of adrenal insufficiency were noted before the age of 2 months, and glucocorticoid and mineralocorticoid replacement was initiated at the time of diagnosis of primary adrenal insufficiency. In late-onset X-linked adrenal hypoplasia congenita, these signs may first present in adulthood, and in such cases, the clinical phenotype is mild [7]. The differential diagnosis of salt-losing primary adrenal insufficiency in male patients includes congenital adrenal hyperplasia, such as 21-hydroxylase deficiency, congenital adrenal lipoid hyperplasia and 3β-hydroxysteroid dehydrogenase deficiency. In adrenal hypoplasia congenita, the adrenal glands lack the adult cortical zone, while they have a normal fetal adrenal cortex, and consequently show normal external male genitalia. In addition, they show different modes of inheritance and have normal or low levels of cortisol precursors, such as 17-OHP.
Patients with adrenal hypoplasia congenita may present entirely absent or arrested pubertal development because of hypogonadotropic hypogonadism. The attainment of secondary sexual characteristics is achieved with testosterone replacement. However, they usually show impaired fertility due to azoospermia or oligospermia despite treatment with gonadotropin therapy [9]. Interestingly, precocious puberty has also been reported in adrenal hypoplasia congenita patients with DAX-1 mutations in rare cases [12-14]. In gonadotropin-independent precocious puberty, high ACTH levels may stimulate the Leydig cells. However, the mechanism of precocity remains unclear, especially in patients with central precocious puberty. In heterozygous females, delayed puberty or adrenal insufficiency can sometimes occur because of skewed X-inactivation.
In the present case, targeted exome sequencing revealed a missense mutation (c.881T>C, p.L294P) in NR0B1 located in the putative ligand-binding domain, which has not been previously reported, although a similar mutation was found in Japan [15]. A likely pathogenic variant according to the American College of Medical Genetics and Genomics guideline was discovered and predicted to be damaging by SIFT and Polyphen-2, and clinical manifestations were compatible with X-linked adrenal hypoplasia congenital [16]. Most missense mutations of the DAX-1 gene are in the ligand-binding domain, as in this case. We hypothesize that this change altered the function of the protein, as the ligand-binding domain is a crucial region, but its function must be further validated in the future.
In conclusion, we report a novel mutation in the NR0B1/DAX-1 gene in a patient with adrenal hypoplasia congenita. Our patient was first diagnosed with adrenal insufficiency at the age of 2 months, and he later manifested the absence of sexual characteristics. Although hormonal tests were performed, the disease etiology was unclear until a genetic analysis was performed. Because a correct and early diagnosis is important for long-term treatment in terms of endocrine and reproductive function and genetic counseling, the possibility of a DAX-1 mutation must be considered in male patients with adrenal insufficiency if a genetic diagnosis has not been reached. Monitoring pubertal development during follow-up is also helpful.
Notes
Ethical statement
This study was approved by the Institutional Review Board of Gangnam Severance Hospital (approval number: 3-2020-0120). The authors have received written informed consent from the patient to use the data and the clinical picture for publication purposes.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.