Role of obesity-induced inflammation in the development of insulin resistance and type 2 diabetes: history of the research and remaining questions
Article information

Abstract
The prevalence of obesity has increased alarmingly both worldwide and in Korea. This has also dramatically increased the prevalence of chronic obesity-associated diseases, including type 2 diabetes (T2D). Extensive studies on the molecular etiology of T2D have revealed several potential mechanisms by which obesity induces the development of insulin resistance and T2D. One of these is low-grade chronic inflammation. Studies hinting at the existence of this phenomenon were first published about 30 years ago. Ten years later, several seminal papers confirmed its existence, which then led to a rapid and massive escalation of research in this field. Today, the notion that obesity-induced inflammation mediates T2D is now well-accepted. This paper will review the key developments in this field, including the discovery that obesity-induced inflammation and insulin resistance is mainly regulated by adipose tissue-resident immune cells, particularly those in visceral adipose tissue. This review further details the research areas, including (1) the obesity-related factors that induce adipose tissue macrophage (ATM) inflammation, (2) the precise effector functions by which adipose tissue immune cells promote insulin resistance, (3) whether there are early immunological events that have an outsize effect on later events and could be targeted to arrest the development of insulin resistance, (4) the roles played by nonimmunological functions of ATMs and other immune cells, and (5) whether there are noncanonical immune responses to obesity (i.e., immune responses that are unique to obesity and cannot be detected by following the discoveries in the classical immunity field).
Highlights
We will review recent progresses for the roles of obesity-induced inflammation in the development of insulin resistance and type 2 diabetes. In particular, we will focus on the regulation of adipose tissues in these processes.
Introduction
In recent decades, the prevalence of obesity has risen dramatically all over the world, including in Korea. This has been accompanied by an equally dramatic increase in a key obesity-associated disease, type 2 diabetes (T2D). For example, in Korea, the prevalence of T2D in 2018 was 14.4% [1] and is projected to continue to increase. Furthermore, both obesity and T2D have been implicated as underlying pathogenic conditions that promote the development and/or aggravation of other diseases, including cardiovascular diseases (CVDs), hypertension, and hyperlipidemia; indeed, mirroring the rise in obesity, the prevalence of these obesity-associated diseases have increased sharply in the last decades as well. These changes have significantly increased the cost of treating T2D or T2D-associated diseases, thus burdening national health care systems all over the world. These costs are also expected to rise dramatically over the next decades.
At present, T2D/T2D-associated diseases are treated with a panoply of prescription medicines. In particular, the last 2 decades have seen the development of new treatments, including several regimens that target novel avenues such as the gut hormone incretin and the glucose-reabsorbing sodium-glucose cotransporter (SGLT)-2. Bariatric surgery has also become more widespread. However, despite the prolific use of these new treatments and other, more established treatments, the prevalence of T2D/T2D-associated diseases—and the death rates due to these diseases—continue to rise. This points to a strong need for new treatments for T2D, particularly ones that target the molecular mechanisms that drive the development of insulin resistance and T2D. This highlights the importance of further investigations into these mechanisms, which remain incompletely understood.
Treatments for T2D
The current treatments for T2D can be categorized on the basis of the 3 stages with which diabetes starts and then progresses, namely, obesity, insulin resistance, and T2D (i.e., hyperglycemia) (Fig. 1). Below, we will first discuss the treatments for obesity (the first stage), then the treatments for T2D (the third stage), and finally the treatments for insulin resistance (the second stage).
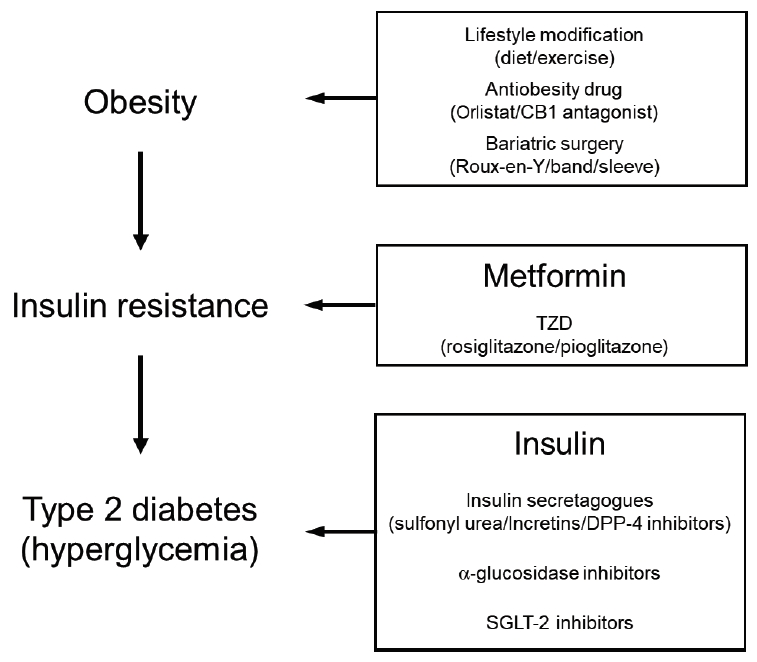
Treatments for type 2 diabetes. CB1, cannabinoid 1; TZD, thiazolidinedione; DPP-4, dipeptidyl peptidase-4; SGLT-2, sodium glucose cotransporter-2.
1. Treatments targeting obesity
Targeting obesity is potentially the most effective treatment regime since it can improve all obesity-associated diseases, including T2D. The most conventional treatment to reduce body weight, including obesity, is to make lifestyle modifications like dietary changes and increasing regular exercise. While this treatment has multiple health benefits, it is strongly limited by the difficulties patients face in long-term compliance with strict diet and exercise programs. Hence, most patients who participate in such lifestyle modification programs return to their original weight within 6 months of finishing the program. As a result, their glycemic levels return to pre-program levels as well [2].
Therefore, there have been multiple attempts to develop drugs that induce weight loss. However, many of these drugs, including fenfluramine/phentermine (fen-phen) and cannabinoid 1 receptor antagonists, have significant and sometimes lethal side effects and have been withdrawn from the market. One drug currently available on the market is orlistat, which prevents fat absorption by inhibiting lipase. However, orlistat is only effective when combined with lifestyle modifications and even then its effects are relatively modest: on average, orlistat in combination with lifestyle changes causes 2–3 kg weight loss [3]. Moreover, the drug induces significant gastrointestinal side effects, in particular steatorrhea (loose oily stools).
Another approach is bariatric surgery. Recent advances have considerably increased its safety, causing bariatric surgery to be adopted worldwide as a solution to obesity. There are several different procedures, the most common being Roux-en-Y and gastric band surgeries. Bariatric surgery acts by decreasing the size of the stomach and/or by blocking food absorption, thereby reducing the volume of food intake. It is well-documented that it leads to substantial long-term weight loss and improvements in T2D/T2D-associated diseases. However, how bariatric surgery improves T2D is not yet fully understood because its effects on T2D precede the loss of body or fat weight. This suggests that mechanisms other than weight loss also underlie the beneficial effect of bariatric surgery on T2D. Possible mechanisms may be upregulation of incretins and changes in the gut microbiome. However, bariatric surgery has several limitations. In particular, it is not recommended for all overweight/obese patients. In the United States, the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases recommend bariatric surgery only for patients whose body mass index (BMI) is 40 kg/m2 or more, or 35 kg/m2 or more if the patient also has a serious health problem (including T2D or heart disease). Patients with a BMI of 30 kg/m2 or more who also have a serious health problem may qualify for gastric band surgery (but not other bariatric surgical modalities). In addition, after surgery, the patients must pay close attention to their diet: the kinds of foods that patients can eat are highly limited, the portions must be small, and micronutrients must be carefully supplemented. Thus, bariatric surgery is quite a drastic approach to treating obesity and should be seen as the last resort for untreatable obesity and T2D.
2. Treatments targeting T2D
Most of the drugs used to treat T2D (the hyperglycemic stage) focus on lowering glucose levels. Since this stage is characterized by poor or no insulin production due to β-cell failure, the primary treatment regimen aims to increase circulating insulin levels. Hence, insulin, the first drug used to treat T2D (starting in 1921), remains a bulwark in the T2D therapy field. Indeed, of the T2D patients in Korea who are treated with prescription drugs, 9.1% are treated with insulin [1]. Recently, closed-loop insulin delivery systems that come with an insulin pump and a continuous glucose monitoring system are now available on the market. These systems were originally developed to treat type 1 diabetes but have been shown to help T2D patients maintain glycemic control as well.
While exogenous insulin directly compensates for endogenous insulin deficiency, there are also less direct approaches. Specifically, a number of insulin secretagogues that induce insulin secretion by β-cells have been discovered and are now used more often than insulin. A classic example is sulfonyl urea. There are also many new secretagogues that act by increasing the levels of the incretin gut hormones. These drugs include the incretins themselves: an example is glucagon-like peptide-1. Others act by prolonging the half-life of incretins: examples are the dipeptidyl peptidase-4 inhibitors. These drugs are increasingly being prescribed.
The latter category of drugs primarily enhances insulin secretion and thus requires at least some β-cell functionality. Therefore, these drugs cannot be used for patients whose β-cells are completely dysfunctional. Drugs for such patients include those that lower circulating glucose levels. One is the α-glucosidase inhibitors, which block the degradation of polysaccharides to glucose, thereby reducing the absorption of glucose by the small intestine. Another drug class is the SGLT-2 inhibitors, which are the newest drugs on the market. These molecules block the reabsorption of glucose by the kidney: this increases the secretion of glucose into the urine while simultaneously lowering the blood glucose levels. SGLT-2 inhibitors not only improve T2D, they also reduce obesity and improve diabetic complications, including CVD and kidney failure, apparently without adverse side effects [4]. As a result, SGLT-2 inhibitor prescriptions have increased dramatically and there is currently active discussion about whether SGLT-2 inhibitors, like metformin, should be recommended as a primary monotherapy for T2D.
3. Treatments targeting insulin resistance
Since the consequences of full-blown T2D, which are often initially hidden, are unlikely reversible, the most ideal step for treating or preventing the development of T2D may be at the insulin-resistant (prediabetic) stage (Fig. 1). Insulin resistance is a metabolic disorder in which β-cells and the cells in the muscles, fat, and liver do not respond well to insulin and therefore cannot maintain glucose homeostasis. This increases blood glucose levels, which induce the β-cells to produce more insulin. The β-cells eventually become exhausted and are killed by the high demand on their insulin production, thereby inducing insulin deficiency and T2D. Thus, treatments that improve the insulin sensitivity of the cells in the muscles, fat, and liver can protect β-cells from this vicious cycle; they can also rehabilitate the β-cells that are starting to become dysfunctional. This will prevent the decline into full-blown T2D. One of the advantages of targeting insulin resistance therapeutically is that it can be determined relatively easily by measuring fasting plasma glucose levels (normal level, 100–124 mg/dL) or hemoglobin A1c (HbA1c) levels (normal level, 5.7%–6.4%).
An important drug that increases insulin sensitivity is metformin. While metformin’s most well-known activity is to inhibit the production of glucose by the liver, metformin also improves insulin sensitivity in animal models and humans by targeting other peripheral insulin resistance in muscle and also fat (however, it does not affect β-cell insulin resistance). It is recommended as a first-line monotherapy for T2D and is thus the most widely prescribed monotherapy for this disease: in Korea, 67.4% of T2D patients were prescribed metformin in 2016 [1]. While the exact molecular mechanisms by which metformin exerts this activity have not yet been fully elucidated, possible molecular mechanisms include improving mitochondrial function, activating the protein kinase AMPK, and changing the gut microbiome (e.g., increasing the numbers of Akkermansia muciniphila [5]).
Another class of drugs that improve insulin resistance is the thiazolidinediones (TZDs), which activate peroxisome proliferator-activated receptor protein γ. This protein has diverse roles in adipocytes and non-adipocyte cells and is intimately involved in insulin sensitivity. TZDs improve peripheral insulin resistance and effectively normalize glycemic control. However, they are also associated with severe side effects, including edema (all TZDs), increased CVD incidents (rosiglitazone), and a potential increase in the risk of bladder cancer (pioglitazone). Therefore, some drugs in this class have been withdrawn from the market in several countries or are now sold with restrictions or warnings.
Currently, metformin and the TZDs are the only drugs available for treating the insulin-resistant stage. Additional drugs targeting this stage are needed. To identify these drugs, it is important to determine the molecular mechanisms that underlie the development of obesity-induced insulin resistance. This imperative has driven extensive investigations over the last few decades. While the mechanisms by which obesity induces insulin resistance remain to be fully elucidated, these studies have shown so far that endoplasmic reticulum stress, hyperlipidemia, mitochondrial dysfunction, and changes in metabolites all play key roles. All of these mechanisms could thus serve as potential targets of interventions that arrest the development of insulin resistance. Another very important mechanism that underlies obesity-induced insulin resistance is chronic low-grade inflammation. As will be discussed in the remainder of this review, many studies in animal models and humans support the existence of this pathogenic mechanism. Thus, below, we provide an overview of the research that led to this discovery by discussing seminal studies at various points in the past. Remaining research questions that could aid the development of interventions that block obesity-induced inflammation and the resulting insulin resistance will then be detailed.
Role of obesity-induced inflammation in the development of insulin resistance and T2D
The idea that obesity-induced inflammation contributes to the development of obesity-induced insulin resistance is now well accepted. Yet the history of this concept is quite short: when PubMed is searched with the terms "obesity" and "inflammation," it shows that the first seminal paper on this topic was only published ~30 years ago in 1993 (Fig. 2). This paper was by the Spiegelman group. It was published in Science and, as will be detailed further below, it drew a direct link between insulin resistance and a cytokine that plays important roles in many inflammatory diseases, namely, tumor necrosis factor (TNF)α [6]. This report is largely believed to be the first to suggest a molecular link between inflammation and insulin resistance.

The number of publications in PubMed using keywords as "obesity" and "inflammation." TNF, tumor necrosis factor; T2D, type 2 diabetes; ATMs, adipose tissue macrophages.
After the 1993 Spiegelman study, the field remained quiet for another 10 years until the epidemiological studies of the Ridker group showed that inflammation in the circulation is associated with T2D and our study demonstrated a direct link between NFκB-dependent inflammation and obesity-induced insulin resistance [7,8]. Thereafter, there was a rapid expansion of publications (Fig. 2) and profound development of the field.
1. 1993: Discovery of the effect of TNFα on insulin resistance and T2D
The seminal 1993 study of Spiegelman and colleagues reported that (1) rodent models of obesity and T2D exhibited high levels of TNFα mRNA in their adipose tissue and (2) neutralization of this cytokine in obese fa/fa rats with a soluble chimeric TNFα-receptor increased their peripheral insulin sensitivity [6].
This study not only linked TNFα to insulin resistance, it also complemented an in vitro study that had been conducted by Pekala et al. [9] in 1983. That study showed that when conditioned medium from endotoxin-treated murine peritoneal macrophages was used to treat an adipocyte cell line, it inhibited some functions of insulin. While Pekala et al. [9] did not know the identity of the mediator(s), it was presumed to be a proinflammatory mediator(s). The 1993 Spiegelman study thus suggested that this mediator was probably TNFα.
In 1997, Uysal et al. [10] further provided in vivo evidence supporting the previous study. They showed that deleting TNF or both its receptors (p55 and p75) reduced obesity-insulin resistance in ob/ob mice or mice with high-fat diet (HFD)-induced obesity. A year later, the same group showed that of the 2 TNFα receptors, it is the p55 receptor that mediates the development of obesity-induced insulin resistance in mouse models [11].
These findings discussed above, namely, that TNFα can impair insulin signaling in vitro and in vivo, have since been well established. These models have thus been used extensively to elucidate the molecular mechanisms by which TNFα interferes with insulin signaling. Spiegelman and colleagues showed that when adipocytes are treated with TNFα, it alters the first events after insulin binding to the insulin receptors. Thus, in the absence of TNFα, insulin-bound insulin receptors undergo Tyr autophosphorylation and then phosphorylate insulin receptor substrate (IRS)-1. By contrast, in the presence of TNFα, Ser phosphorylation of IRS-1 occurs. This converts IRS-1 into an inhibitor of the insulin receptors, namely, it downregulates the tyrosine-kinase activity of the insulin-bound insulin receptors. As a result, TNFα treatment decreases the Tyr autophosphorylation of the insulin receptors in adipocytes, thereby blocking insulin signaling [12]. Although the mechanism by which Ser-phosphorylated IRS-1 inhibits the insulin receptors has not been further explored in detail, it is now well known that TNFα induces Ser phosphorylation of IRS-1, notably at Ser 302 and 307, and that this is associated with the status of insulin signaling and insulin resistance in vivo as well as in vitro [13,14]. Other reagents that inhibit insulin signaling have the same effect on IRS-1: these reagents include phorbol 12-myristate 13-acetate and anisomycin, which respectively activate protein kinase C and cJun-N-terminal-kinase. However, it has not yet been fully explored whether these IRS-1 Ser phosphorylation events actually impair insulin signaling; it remains possible that they are simply associated with insulin resistance. In any case, IRS-1 Ser phosphorylation is considered to be an acute response to TNFα.
Notably, we also have evidence that chronic (not acute) exposure of adipocytes to TNFα regulates the transcription of IRS-1: when adipocytes are exposed to TNFα in vitro for prolonged periods (72–96 hours), IRS-1 mRNA and protein levels drop. Prolonged TNFα exposure also reduces insulin-stimulated transport of glucose in adipocytes by decreasing the expression of the insulin-responsive glucose transporter Glut4 [15].
The ability of TNFα to induce the acute and chronic responses described above (i.e., Ser phosphorylation of IRS-1 and transcriptional downregulation of insulin-signaling molecules like IRS-1 and Glut4, respectively) has since also been observed in insulin-resistant/T2D animals and humans.
Notably, 2 studies around this period showed that the adipose tissues of obese humans express higher levels of TNFα protein and mRNA and that weight loss is associated with a reduction in both adipose TNFα levels and insulin resistance [16,17]. Hence, the next question in the field was whether inhibiting TNFα function in humans could improve T2D. This possibility could be readily tested because TNFα inhibitors that effectively treat rheumatoid arthritis had recently been discovered and marketed. Thus, the ability of TNFα inhibitors to improve T2D was tested by several small-scale clinical trials. However, while some trials showed that TNFα improved glycemic control, most did not show any positive results, although some did demonstrate reduced expression of the inflammatory marker C-reactive protein (CRP). However, most of these studies had relatively small sample sizes. Hence, the role of TNFα in the development of T2D and whether inhibiting TNFα can effectively treat T2D remain to be determined [18].
It should be emphasized that the latter studies all focused on one specific proinflammatory cytokine rather than on inflammation per se. This reflects the relatively immature state of immunology at the time. Indeed, nuclear factor-kappa B, which is now known to be the master regulator of inflammation, was only cloned in 1986 [19]; moreover, its regulation by its inhibitor IκBα and the IκBα kinases was first published in 1990 [20] and 1997 [21], respectively. Furthermore, toll-like receptor (TLR)-4, which has since been established as a critical mediator of inflammation, was not identified or cloned until 1996 [22-24]. Nevertheless, the research on the role of TNFα in obesity-induced insulin resistance remains important to this day for 2 reasons. First, it is now well known that TNFα plays a critical role in regulating inflammation in classical immunity settings such as infection and autoimmunity. Thus, it is possible that TNFα contributes significantly to insulin resistance, although this role is unlikely to be as simplistic as was believed in the 1990s. Second, the TNFα studies are the first of a vast series of studies that link inflammation to obesity, insulin resistance, and T2D.
2. 2001: Discovery that inflammation acts as a pathogenic mediator of the development of obesity-induced insulin resistance and T2D
Although many of the studies reviewed in the previous section were published in high-impact journals and were the focus of much discussion, the number of papers on the link between inflammation and obesity did not change dramatically until the early 2000s (Fig. 2). In 2001, an epidemiological study with the Woman's Health Initiative Study cohort led by the Ridker group showed that increased levels of circulating interleukin (IL)-6 and high-sensitivity (hs)CRP predict a high risk of developing T2D in the future [7]. The studies on T2D were an extension of an earlier study on CVD that was also led by the Ridker group; this earlier study showed that circulating levels of proinflammatory mediators (including IL-6 and hsCRP) also predict the risk of CVD in humans [25]. Together, these studies drew a strong link between inflammation and the future development of metabolic diseases such as T2D and CVD. While the epidemiological design of these studies meant that it was not possible to definitively state that inflammation causes these diseases, they were exceedingly important because they suggested that inflammation in general may be an underlying pathogenic mediator of T2D in humans.
Coincidently, in 2001, we published a study in rodent models showing that inflammation directly promotes the development of obesity-induced insulin resistance via an IKKβ/NFκB-dependent pathway. We showed that obesity activates this pathway, and that inhibiting this pathway by knocking out IKKβ or administering inhibitors of this pathway (i.e., high doses of aspirin or salicylates) prevented the development of obesity-induced insulin resistance [8]. Subsequently, this study was translated into a small open-labelled clinical study on the effect of salsalate on insulin resistance and T2D. Salsalate is a dimer of salicylates and has fewer side effects than salicylates or aspirin. We found that high doses of salsalate improved the glycemic control of patients with insulin resistance and T2D; compared to baseline, the drug lowered fasting glucose levels and increased glucose utilization in euglycemic hyperinsulinemic clamps. The drug also decreased NF-κB activity in the circulating immune cells of patients [26].
These preclinical and clinical data together show that IKKβ/NFκB-dependent inflammation directly causes the development of insulin resistance and T2D in animal models and humans and that inhibiting this inflammation improves these diseases. This suggests that anti-inflammatory therapy may be effective against T2D. This notion was then directly tested and confirmed by the Targeting INflammation Using SALsalate in type 2 diabetes (TINSAL-T2D) study series, which comprised 2 multicenter placebo-controlled double-blinded trials on salsalate [27,28]. Both studies showed that compared to placebo, treating T2D patients with salsalate significantly improved their glycemic control, as indicated by HbA1c levels.
After these seminal studies and trials, the number of publications in the T2D field started to rise sharply (Fig. 2).
3. 2003: Discovery that adipose tissue macrophages regulate obesity-induced inflammation
The next question that arose in the field was, "Which tissues/cells regulate the inflammation that appears to drive obesity-induced insulin resistance and T2D?" Initially, focus was on the insulin-responsive cells, namely, the hepatocytes, adipocytes, and myocytes in the liver, adipose tissue, and muscles, respectively. However, when the IKKβ/NFκB pathway was upor downregulated in these cells, the expected changes were not (fully) observed. Thus, when the IKKβ/NFκB pathway was either inhibited or activated in hepatocytes by respectively deleting IKKβ or overexpressing the dominant negative IκBα, or overexpressing constitutively active IKKβ, obesity-induced insulin resistance in the liver improved and worsened, respectively [29,30]. However, the changes in insulin resistance in these mice were mainly due to the respective reduction and elevation of the insulin levels; modulating the IKKβ/NFκB pathway in hepatocytes did not change glucose levels. This disparity was not observed when (as discussed in the previous section) IKKβ was knocked out in the whole body and the mice were fed with an HFD, or HFD-fed mice were treated with salicylate. In these cases, insulin and glucose levels both dropped [8]. Similarly, when constitutionally active IKKβ was expressed in myocytes, severe muscle wasting was observed in the mice and an HFD did not induce insulin resistance [31]. Moreover, when the IKKβ/NFκB pathway was downregulated in adipocytes by deleting IKKβ, it reduced fat weight but paradoxically induced (not suppressed) inflammation in the adipocytes and worsened HFD-induced insulin resistance [32,33]. The effect of deleting IKKβ on adipocyte inflammation was later confirmed by a study in 2016 [32]. These studies led to suspicion that other cells, such as immune cells, regulate the development of obesity-induced inflammation.
This question "Which cells/tissues?" was directly answered by 2 seminal papers in 2003. The Ferrante and Chen groups independently showed that when mice became obese due to genetics or an HFD, the macrophages in the adipose tissue markedly increased in number. Obesity also increased the expression by these adipose tissue macrophages (ATMs) of proinflammatory genes; this elevated inflammation was not observed in the adipocytes of the obese mice [34,35]. Arkan et al. [30] then showed that myeloid cells were important for establishing obesity-induced insulin resistance: when IKKβ was deleted in myeloid cells, the mice were protected from developing obesity-induced insulin resistance. As summarized by several reviews [36,37], numerous other studies then showed that deleting inflammation-related genes in myeloid cells (by using LysM-cre mice) simultaneously improved both obesity-induced insulin resistance and adipose tissue inflammation. These data together support the notion that ATMs are a major player that regulates the development of obesity-induced inflammation and insulin resistance.
Initial characterization of ATMs by Lumeng et al. [38] then suggested that obesity induces ATMs to polarize from M2-type macrophages into M1-type macrophages; while ATMs in lean mice expressed M2 markers (Ym1, Arg1, and IL-10), obesity induced the appearance of new ATMs expressing the M1 marker CD11c+. The M1/M2 categorization of macrophages was originally developed by in vitro studies in the classical immunology field but is now also used in in vivo studies in many other fields, including the immunometabolism field (Table 1) [39,40]. M1 macrophages are considered to be proinflammatory macrophages and were originally defined according to the genetic signatures of bone marrow-derived macrophages (BMDMs) that had been stimulated with interferon-γ and lipopolysaccharide. M2 macrophages were defined according to the BMDM gene signatures that were stimulated by IL-4 or IL-13. M2 macrophages are generally considered to be anti-inflammatory cells but also play a role in wound healing and remodeling. After the study by Lumeng et al. [38], various studies showed that obesity dramatically increases the frequencies of CD11c+ ATMs in the epididymal/visceral adipose tissues from both rodent models and humans, respectively. Therefore, the general consensus at the time was that the ATMs that regulate the development of obesity-induced inflammation are proinflammatory CD11c+ M1-type ATMs.

M1 and M2 macrophages
However, this notion, and the idea that the M1/M2 dichotomy neatly characterizes the link between proinflammatory ATMs and obesity-induced inflammation, was subsequently challenged by multiple studies. First, a gene expression analysis showed that sorted CD11c+ and CD11c- ATMs did not actually differ significantly in terms of inflammatory profiles. The same study showed by fluorescence-activated cell sorting (FACS) analysis that obesity also increased the frequency of ATMs expressing the M2 marker CD206; notably, this rise in CD206 expression was also observed in CD11c+ ATMs [41]. Second, gene expression analyses of sorted total ATMs from lean and obese animals showed that obesity increased the expression of not only M1/proinflammatory markers (including TNFα and IL-1β) but also M2/anti-inflammatory markers such as Arg1 and IL-10. The same study also found that compared to lean ATMs, the NFκB target genes in obese ATMs were not always upregulated: many of these genes were in fact downregulated [42]. Thus, obesity clearly upregulates specific pro- and anti-inflammatory mediators but in a pattern that does not fit tidily into the M1/M2 classification [42]. Nevertheless, the primary finding of Lumeng et al. [38], namely, that obesity upregulates CD11c+ ATMs, has been confirmed repeatedly in both animals and humans. However, the prodiabetic functions of these specific cells are still not fully understood.
Moreover, it has recently been shown that ATMs play much more diverse roles in the regulation of metabolic processes than just regulating inflammation. These roles include lipid metabolism [41,43], thermogenic regulation via brown fat differentiation/function [44,45], and adipose tissue fibrosis [46,47], all of which can also shape the development of obesity-induced insulin resistance.
Thus, ATMs are not only the most abundant adipose tissue immune cells, they play very important roles in the development of obesity-induced insulin resistance and T2D by regulating inflammation along with lipid metabolism, thermogenesis, and/or adipose tissue fibrosis. The studies of the Ferrante and Chen groups in 2003 caused ATMs to become a major focus of research on T2D, with the result that the numbers of publications in the T2D field started to increase dramatically (Fig. 2).
4. 2009: Discovery of the roles of other adipose tissue immune cells on obesity-induced inflammation
By 2009, studies in the classical immunity field had started to show that macrophage regulation was very complex and involved many other immune cell types. This caused many fields of research, including the T2D field, to shift focus from the role of macrophages in inflammation to determining how other immune cell types regulated inflammation. In 2009, Nature Medicine published a series of 4 papers that reported the roles of CD4 T cells, CD8 T cells, Tregs, and mast cells in the development of obesity-induced inflammation and insulin resistance [48-51]. They all utilized knockout mouse models and cell-specific depletion and/or reconstitution of target cells; all also analyzed the effects of these changes on adipose tissue inflammation. Thus, to determine the role of CD4 T cells, the authors used RAG knockout mice, which are completely deficient in T and B cells. Metabolic phenotype analyses showed that when fed an HFD, these mice exhibited increased blood insulin levels and adipose tissue inflammation compared to wildtype. When the mice were reconstituted with CD4 T cells isolated from normal chow-fed wildtype mice and were then fed an HFD, their insulin sensitivity was better; reconstitution was also associated with lower adipose tissue inflammation. This initially suggested that CD4 T cells play a protective role in the development of obesity-induced adipose tissue inflammation and insulin resistance. However, this finding was complicated by the fact that compared to wildtype mice, the RAG knockout mice had higher body and fat weights; moreover, reconstitution with CD4 T cells normalized these weights. Hence, it is not clear whether CD4 T cells regulate the development of obesity-induced insulin resistance directly; it is also quite possible that CD4 T cells regulate it only indirectly by controlling obesity (which then regulates insulin resistance in a secondary fashion). Similarly, the study on mast cells showed that although mast cells appeared to protect against obesity-induced insulin resistance, mast-cell knockout also regulated body and fat weights [51]. By contrast, the studies on Tregs and CD8 T cells showed that both of these cell types protected against obesity-induced adipose tissue inflammation and insulin resistance but did not regulate obesity [48,50]. Therefore, the latter 2 cell types directly regulate the development of obesity-induced insulin resistance.
Later studies investigated the roles of most other adipose tissue immune cell types in the regulation of obesity-induced inflammation and insulin resistance. These studies examined cell-specific knockout mouse models and their reconstitution with the target cells, target-cell depletion using antibodies, and/or expansion/activation of the target cells. The roles that were identified aligned closely with their traditional inflammatory roles, as determined in the classical immunity field. Thus, cells that are proinflammatory cells in the classical immunity field, including dendritic cells (DCs), neutrophils, Th1 cells, Th17 cells, γδT cells, B cells, natural killer (NK) cells, and innate lymphoid cells (ILC)1, were found to induce both obesity-induced inflammation and insulin resistance [49,52-58]. By contrast, cells that are anti-inflammatory cells in the classical immunity field, including Breg, eosinophils, and ILC2, suppressed both obesity-induced inflammation and insulin resistance [59-61]. Curiously, one cell type, invariant NK T (iNKT) cells, exhibited diametrically different results in different labs. Some labs showed that iNKT cells play a detrimental role in obesity-induced insulin resistance, some showed the opposite, and others showed that iNKT cells did not regulate insulin resistance in either direction. It remains unclear why different labs show these dramatic differences despite the fact that most of the labs used the same mouse lines (reviewed in [37]).
It should be noted that when the numbers of some of these immune cell types were altered by genetic knockout/reconstitution, expansion, and/or depletion, the changes also altered the body and fat weights of normal chow-fed animals. These cells include Th2 cells, DCs, neutrophils, eosinophils, ILC2 [49,51-53,62], and in some cases, iNKT cells (reviewed in [37]). Thus, it is unclear whether these cells regulate obesity-induced insulin resistance directly or only secondarily. Indeed, recent studies suggest that some immune cell types, namely ILC2, Th2 cells, and eosinophils, help regulate the development of beige fat and that these activities are associated with changes in body and fat weights. Hence, adipose tissue immune cells can regulate obesity-induced insulin resistance by directly shaping adipose tissue inflammation and/or by indirectly affecting it via their obesity-regulating activities.
Regardless of how adipose tissue immune cells regulate obesity-induced adipose tissue inflammation, their effects all share the following common features. First, their regulatory activities are restricted or more evident in epididymal fat (rodent models) and visceral fat (humans) compared to subcutaneous fat. Second, activating or suppressing a pro- or anti-inflammatory immune cell type is associated with corresponding changes in ATM inflammation. For example, depleting NK cells suppresses the expression of proinflammatory/M1 genes while simultaneously increasing their anti-inflammator y M2 gene expression in FACS-sorted ATMs. Expansion of NK cells has the opposite effect on the ATM gene profile. These changes are associated with corresponding changes in the inflammatory profile of total adipose tissue [57]. Third, most of the adipose tissue immune cells exhibit distinctive immunophenotypes compared to the equivalent cell types in other tissues such as the liver, spleen, and blood. For example, adipose tissue CD4 and CD8 T cells, Tregs, and B cells all express unique T-cell or B-cell receptor repertoires [48,49,56]. Another example is that obese ATMs express very high levels of CD11c despite the fact that it is a classical marker for DCs in most other tissues. Similarly, obese (and therefore activated) NK cells produce large amounts of proinflammatory cytokines such as TNFα but have low cytotoxic activity [57,63]; in more classical immunity settings (such as infection), these NK cell activities are more balanced.
These studies together have led to a general consensus that obesity disrupts the immune homeostasis between pro- and anti-inflammatory immune cells in the visceral fat and that this promotes systemic insulin resistance in the liver and muscle (Fig. 3).
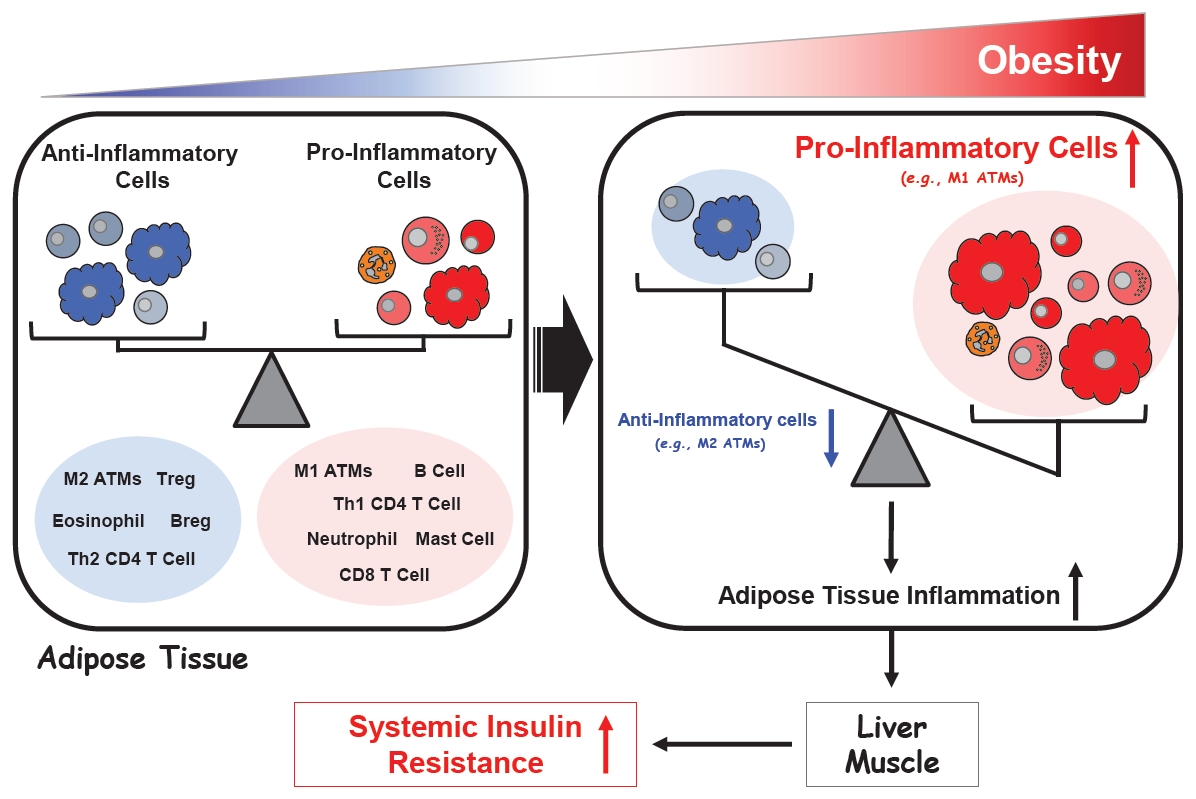
Regulation of adipose tissue inflammation in obesity.
5. 2017: A large clinical trial on an anti-inflammatory therapy in CVD and T2D patients: the CANTOS trial
The studies discussed above established that inflammation plays an important role in the development of obesity-induced insulin resistance. However, the critical question, "Can anti-inflammatory interventions treat T2D in humans?" remained to be fully answered. Therefore, there was considerable excitement in the field when the CANTOS trial was published in 2017. This 48-month-long, randomized, placebo-controlled, double-blinded trial tested the ability of repeated 3-month injections of an anti-IL-1β monoclonal antibody (canakinumab) delivered at a dose of 50, 150, or 300 mg to improve atherosclerotic disease in more than 10,000 subjects with previous myocardial infarction and systemic inflammation as defined by circulating hsCRP levels of >2 mg/L [64]. The latter inclusion criteria, which are highly specific, should be noted because they will be discussed further below. The primary study end point was incidence of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death. There were also multiple secondary end points, including the incidence of diabetes. Significantly, canakinumab markedly lowered the circulating hsCRP levels regardless of dose but had no effect on circulating lipid levels. Moreover, all 3 doses lowered the recurrence rates of cardiovascular events, although only the middle dose (150 mg) met the prespecified primary endpoint. However, compared to placebo, all 3 doses were also associated with a relatively small but significantly higher incidence of fatal infections (0.31 vs. 0.18 events per 100 person-years, P=0.02). Nevertheless, canakinumab did not increase all-cause mortality rates. Interestingly, canakinumab significantly decreased the reports of arthritis, gout, and osteoarthritis and reduced lung cancer incidence and mortality [65]. These studies conclusively show that inhibiting inflammation, specifically IL-1β, in patients with atherosclerosis improves CVD outcomes, especially myocardial infarction recurrence rates. Thus, canakinumab may be a new treatment for CVD.
In a subsequent analysis of the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) trial data, the participants were divided according to whether they were diabetic, prediabetic, or had normal glucose levels at baseline (as determined by HbA1c levels). As expected for a population with atherosclerosis, 40.3%, 49.3%, and 10.4% of the patients fell into these subgroups, respectively. Canakinumab had no long-term (>9 months) effect on the fasting blood glucose or HbA1c levels in any of the 3 subgroups. Moreover, when the patients who were prediabetic at baseline were examined for the development of new-onset diabetes during the study, canakinumab treatment was found to have no effect on the incidence of new-onset diabetes (the rates per 100 person-years in the 4 study arms ranged from 4.1 to 4.4; log-rank P=0.84) [66].
This result was contrar y to expectations and clearly disappointing to the field. However, since the CANTOS trial was designed to detect the effect of canakinumab on CVD outcomes, the T2D outcomes should be interpreted carefully. In particular, the cohort in the CANTOS trial may not be suitable for testing the effect of an anti-inflammatory intervention on T2D for 2 reasons. First, all patients had a preexisting condition (i.e., CVD that had led to previous myocardial infarction). This comorbidity complicates data interpretation; as a result, T2D-focused clinical trials generally exclude subjects with preexisting conditions, including CVD. Second, all subjects had high circulating levels of hsCRP (>2 mg/L), as mandated by the eligibility criteria. Although hsCRP has been clinically used to determine systemic inflammatory status, and high hsCRP levels are associated with a high risk of T2D [66], the role that this protein plays in the development of T2D is still not fully understood. Moreover, T2D clinical trials generally do not use hsCRP levels as an eligibility criterion and therefore include subjects with hsCRP <2 mg/L. A third point that cautions against direct extrapolation of the CANTOS trial results to the T2D field is that the CANTOS trial only tested the effect of inhibiting a single proinflammatory mediator, IL-1β. It is quite possible that the pathogenic inflammation that drives T2D is not (or not strongly) mediated by IL-1β or secondary inflammatory responses to this cytokine.
These points are exemplified by the TINSAL-T2D trials that were published in 2010 and 2013 [27,28]. These trials recruited subjects with T2D but excluded those with a history of many other chronic inflammatory diseases, including unstable CVD. Moreover, these trials tested salsalate, which has much broader effects on inflammation. Significantly, these trials showed that unlike IL-1β inhibition, salsalate consistently lowered HbA1c levels in T2D patients [27,28]. This supports the notion that broader suppression of inflammation may improve T2D. In relation to this, in 2016, a TINSAL trial also tested the effect of salsalate on CVD outcomes in overweight and obese patients who were using statins [67]. The findings of this trial were very similar to the TINSAL-T2D findings and contrary to the CANTOS findings. Thus, unlike CANTOS, the TINSAL-CVD study found that the study drug (salsalate) had no effect on CVD outcomes. Moreover, unlike CANTOS but like TINSAL-T2D, the TINSAL-CVD study showed that the study drug (1) improved glycemic control, (2) had no effect on hsCRP levels, and (3) reduced the counts of circulating leukocytes, neutrophils, and lymphocytes.
These observations suggest that although inflammation plays an important role in the development of both CVD and T2D, and anti-inflammatory interventions may be a new way to treat both diseases, the biological and pathophysiological nature of the inflammation in these diseases may be very different. Hence, different inflammation-suppressing approaches may be needed in CVD and T2D.
6. 2020: New questions
The notion that obesity-induced inflammation plays an important role in the development of insulin resistance and T2D was established in 2001. Since then, significant progress has been made in this field, as evidenced by the dramatic increase in the numbers of publications after 2001 (Fig. 2). However, there are still numerous questions that need to be addressed by both preclinical basic research and clinical studies, as follows:
1) How are immune cells regulated by obesity?
It is now well established that obesity activates proinflammatory immune cells while suppressing anti-inflammatory immune cells. However, the molecular mechanisms that underlie this pattern remain very unclear. The first question is, which initial mediators activate/suppress immune cells in obesity? An early study suggested that free fatty acids may activate macrophages via TLR4 [68]. However, a later study disputed the role of TLR4: it showed that deleting TLR4 in macrophages did not affect the development of obesity-induced inflammation and insulin resistance in mice [69]. In addition, when MyD88, a universal signaling adaptor molecule for TLRs, was deleted, the mice even showed a mild increase in insulin resistance [70]. Hence, the initial mediators that regulate immune cell activities in obesity remain contested. Potentially, they include lipids, metabolites, cytokines/hormones, and/or environmental stress.
Another question is, which tissues are inflamed by obesity? A recent study showed clearly that of the tissues examined (fat, liver, muscle, and spleen), only epididymal fat is inflamed by obesity [57]. However, other as yet untested tissues/organs, including the circulatory system, may also be affected by obesity. Understanding the global immune response of the whole body to obesity would provide a more holistic view of the diseases it causes.
It is also not clear whether there are seminal immunological events that drive obesity-induced inflammation and insulin resistance, and when in the disease process they occur. Classical immunity studies show that key immunological events that trigger a gradual slide into a diseased state (e.g., autoimmunity) can occur well before signs of pathology are observable. However, most studies on T2D-related inflammation focus only on later stages of obesity, such as those in mice that have been fed a 60% HFD for 12 weeks: at this point, insulin resistance is well established. There are only a few studies on early changes after starting an HFD; while most other cells (e.g., macrophages, Treg, and NK cells) play a role in the later stages of obesity (>12 weeks on an HFD), neutrophils and CD8 T cells appear to regulate HFD-induced inflammation at the early stages of the disease (1–4 weeks on an HFD) [50,53]. However, studies that systematically dissect how and which immune cells are regulated during the progression of obesity remain lacking.
2) How does crosstalk between immune cells participate in obesity-induced inflammation?
Classical immunity studies show that the regulation of immune cell homeostasis involves considerable crosstalk between different immune cell types. The role of immune cell crosstalk in the regulation of obesity-induced inflammation and insulin resistance has been poorly addressed to date because most studies focused on a single immune cell type. Recently, however, several studies have started to examine the crosstalk of different immune cells in adipose tissue, with a particular focus on ATMs. For example, we showed that NK cells directly control the recruitment of ATMs into the adipose tissue during an HFD; these cells also directly control the expression of inflammatory-mediator genes in ATMs [57]. However, whether (and how) NK cells regulate immune cells other than ATMs in obesity has not been assessed.
Some immune cell crosstalk involves regulatory cells directing the behavior of effector cells. Many different immune cells can be categorized as effector cells and regulatory cells. The communication from the regulatory cells is mainly mediated by cytokines. However, at present, it remains unclear which cells are targeted by these regulatory cytokines and how this affects immune homeostasis in lean and obese conditions.
Once directed by a regulatory cell, the effector cell exerts its activities on other cells, thus creating a cascade of effector responses. It is unclear how the key effector cells in obesity, namely, ATMs, regulates the development of obesity-induced insulin resistance. For example, the mediators involved and the cellular targets are incompletely understood. Even less well understood is how other effector cells in obesity exert their effects.
In addition, immune cell effector functions are not restricted to inflammatory roles: most immune cell types consist of many different subpopulations, some of which have functions that do not relate directly to immunity. For example, macrophages also play an important role in cutaneous wound healing; in the early inflammatory stage of this process, macrophages induce fibroblasts to lay down extracellular matrix to fill the defect. Later, the macrophages change their activities to induce tissue remodeling that strengthens the dermis. If the inflammatory stage is abnormally prolonged and the remodeling stage is inadequate, abnormal wound healing and fibrotic scarring ensues [71]. Obese adipose tissues often show fibrosis, which suggests that the local macrophages have promoted excessive fibroblastic activities in the fat. Since most of the studies on obesity-induced inflammation and insulin resistance have focused on the inflammatory roles of adipose tissue immune cells only, little is known about the roles of their other activities. This leads to many questions, such as, do the ATMs in obesity also play an important role in fibrosis? Moreover, since macrophages also engulf fat and can become lipid-laden foam cells in obesity, do they have any other as yet unresolved functions such as lipid metabolism?
Answering all of these questions will help us to understand how immune cells in obesity regulate and exert inflammatory and other non-inflammatory activities and how this then controls the development of insulin resistance and T2D.
3) Questions relating to anti-inflammatory treatments for human T2D patients
The ultimate goal of studying obesity-induced inflammation is to find new and effective treatments for patients with insulin resistance/T2D. However, the clinical trials on anti-inflammatory drugs to date are limited and the 2 large trials (CANTOS and TINSAL-T2D) showed disparate results [27,66]. This suggests that more preclinical studies are needed to more fully understand the nature of obesity-induced inflammation, thereby helping to identify possible therapeutic targets.
An ongoing concern regarding any anti-inflammatory intervention is that it could prevent the patient from mounting appropriate immune responses to infectious agents. Indeed, the CANTOS trial showed that canakinumab significantly increased the incidence of sepsis [64]. However, this concern could be tempered in relation to T2D by the fact that the obesity-induced immune state differs in key ways from the immune state that is seen in classically studied disorders such as genetic, infectious, and autoimmune diseases and cancer. In particular, obesity-induced inflammation is chronic and low-grade and, as indicated by the studies on TLR4/MyD88, may involve noncanonical inflammation-related pathways. Thus, it may be possible to identify an intervention that targets T2D inflammation without harming immune responses to infectious agents. However, identifying such obesity/T2D-specific targets is currently hampered by the fact that the T2D field generally follows the discoveries in the classical immunity field: a common research strategy is to take new findings from the classical immunity field and test them in the obesity setting. While this is a reasonable approach in many regards, the novel characteristics of obesity-induced inflammation should also be investigated independently without necessarily referring to classical immunity research (which may not always be relevant to the immunometabolism field and could even be a red herring that wastes research resources).
An ongoing concern regarding any anti-inflammatory intervention is that it could prevent the patient from mounting appropriate immune responses to infectious agents. Indeed, the CANTOS trial showed that canakinumab significantly increased the incidence of sepsis [64]. However, this concern could be tempered in relation to T2D by the fact that the obesity-induced immune state differs in key ways from the immune state that is seen in classically studied disorders such as genetic, infectious, and autoimmune diseases and cancer. In particular, obesity-induced inflammation is chronic and low-grade and, as indicated by the studies on TLR4/MyD88, may involve noncanonical inflammation-related pathways. Thus, it may be possible to identify an intervention that targets T2D inflammation without harming immune responses to infectious agents. However, identifying such obesity/T2D-specific targets is currently hampered by the fact that the T2D field generally follows the discoveries in the classical immunity field: a common research strategy is to take new findings from the classical immunity field and test them in the obesity setting. While this is a reasonable approach in many regards, the novel characteristics of obesity-induced inflammation should also be investigated independently without necessarily referring to classical immunity research (which may not always be relevant to the immunometabolism field and could even be a red herring that wastes research resources).
Another concern relates to the potentially high cost of developing biological anti-inflammatory treatments such as canakinumab. The developers of such treatments must pass the development costs on to the patients, which could make such treatments unaffordable. This practical consideration should also be taken seriously when searching for T2D interventions.
Closing remarks
While studies on the role that obesity-induced inflammation plays in the development of insulin resistance and T2D only started around 30 years ago, the progress in this field has been dramatic. This reflects the vast promise of this area in terms of finding an effective intervention for insulin resistance/T2D, which has become an enormous and growing public health issue on a global scale. However, further preclinical and clinical research is clearly needed to further advance the field to the point that effective anti-T2D drugs that act by suppressing obesity-induced inflammation and/or other key immune cell activities can be identified.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by Soonchunhyang University and the Regional Leading Research Center (RLRC) grant of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT), Kore, to J.L (NRF-2019R1A5A8083404).