Dizygotic twin sisters with normosmic idiopathic hypogonadotropic hypogonadism caused by an FGFR1 gene variant
Article information

Abstract
Isolated hypogonadotropic hypogonadism (IHH) is a rare genetic disorder that is clinically and genetically heterogeneous. It is characterized by absent or incomplete pubertal development owing to an isolated defect in the production, secretion, or action of gonadotropin-releasing hormone. The incidence of IHH is estimated at 1:30,000 in males and 1:125,000 in females. Although the vast majority of IHH cases are sporadic, some X-linked recessive, autosomal dominant, and autosomal recessive modes of inheritance have been described. IHH can be classified into Kallmann syndrome with anosmia and normosmic IHH. Here, we report dizygotic twin sisters with normosmic IHH who showed short stature and absence of puberty as a result of a variant of the FGFR1 gene. They had a normal sense of smell, and brain magnetic resonance imaging (MRI) showed well-defined olfactory bulbs. The older sister and the twins' mother had cleft palate, while the younger sister did not. The mother had menarche at the age of 16 years after hormonal replacement owing to delayed puberty. Molecular analysis of the FGFR1 gene identified a missense variant c.874C>G (p.His292Asp) in the twins and their mother. Herein, we described the clinical heterogeneity observed in the 2 affected twins who carry an identical variant in the FGFR1 gene. Further studies of the effects of modifier genes and epigenetic factors on the expression of FGFR1, as well as the various clinical manifestations of its mutations, are warranted.
Introduction
Idiopathic hypogonadotropic hypogonadism (IHH) is characterized by delayed or absent sexual development owing to low levels of sex steroids. Low or inappropriately normal levels of luteinizing hormone (LH) as well as follicle-stimulating hormone (FSH) are common, but no anatomical abnormalities are observed in the hypothalamic-pituitary gonadal axis [1]. Patients with IHH have a deficit in hypothalamic gonadotropin-releasing hormone (GnRH) production or function [2]. IHH is classically divided into 2 major categories: IHH with anosmia or hyposmia (Kallmann syndrome [KS]) and IHH with normal olfaction (normosmic IHH [nIHH]) [3]. Approximately 60% of patients with IHH present with KS, while the remaining 40% have nIHH [4]. Patients with KS may have additional phenotypic abnormalities including craniofacial defects, neurosensory deafness, digital abnormalities, unilateral renal agenesis, and neurological defects, whereas those with nIHH usually do not exhibit any other malformations [5].
IHH is a clinically and genetically heterogeneous condition that can be sporadic or familial and the latter type is often inherited as either an X-linked or autosomal trait [6]. Only 30%–50% of all patients with IHH have currently known genetic defects [1]. However, gene mutations have been identified in only 2%–16% of patients with nIHH, the most frequent of which are inactivating mutations of the GnRH receptor (which are more prevalent in patients with familial disease) [7]. The incidence of this disease is 1:125,000 and 1:30,000 among female and male populations, respectively [8]. Although the reason for this marked discrepancy between the sexes is not known, the incidence of the disease is probably underestimated in females [9] because there typically is no clinical manifestation of IHH among girls before the early teenage years [3].
We report a case of dizygotic twin girls with nIHH who presented with short stature and delayed puberty. The clinical and genetic aspects of these patients are reviewed and discussed, as are the therapeutic strategies we followed.
Case reports
Dizygotic twin sisters who were 11.5 years of age were referred to the endocrinology clinic of our hospital for short stature and absence of puberty. They were born from nonconsanguineous parents. Their mother was 31 years old at the time of their birth; she had been treated with hormone therapy because of delayed puberty, and was 16 years of age at the time of menarche. She had a history of cleft palate that had been surgically corrected, but had no anosmia. After undergoing artificial insemination because of her paramenia, she gave birth to the dizygotic twins at a gestational age of 38 weeks. The mother's height was 166 cm and the father's height was 173 cm. Their daughter's midparental target height was 163 cm.
1. Case 1
The first of the twin girls had a birth weight of 2.35 kg (<3rd percentile) and had no remarkable prenatal or perinatal history. She was born with a cleft palate that was corrected surgically. On physical examination at the age of 11.5 years, her height and body weight were 136.1 cm (-1.5 standard deviation score [SDS]) and 31.7 kg (-1.1 SDS), respectively (Table 1). Her breast and pubic hair stages were Tanner I. There was no evidence of nystagmus, cerebellar ataxia, mirror movement, sensorineural hearing loss, or anosmia. Her karyotype was 46,XX, but her bone age was 2 years behind her chronological age. At the age of 15 years, she had undergone no pubertal changes and her growth velocity was 5–6 cm/yr, which was normal for prepubertal girls. A combined pituitary stimulation test was performed when she was 15 years old; basal hormonal tests revealed low serum estradiol (<5.0 pg/mL) as well as suppressed LH (0.8 IU/L) and FSH (2.2 IU/L) levels. When undergoing a GnRH stimulation test, her LH and FSH peaked at 15.9 IU/L and 11.8 IU/L, respectively. Her anterior pituitary functions as well as thyroid functions were normal. Olfactory function was examined using the Sniffin' Sticks test (KVSS, Korean version) [10]. MRI revealed a normal hypothalamic-pituitary region with an intact olfactory bulb and sulci. Pelvic ultrasonography revealed an infantile uterus and small ovaries. Her bone mineral density (BMD) (corrected for bone age) was reduced; her L1–4 spine BMD z-score was -3.03.

Clinical characteristics of dizygotic twins with normosmic idiopathic hypogonadotropic hypogonadism
Clinical exome sequencing for genetic analysis revealed a novel heterozygous missense variant c.874C>G (p.His292Asp) in FGFR1 that was identified through high-throughput sequencing, exon captures, bioinformatics analysis, and clinical database analysis. Validation using Sanger sequencing and pedigree verification (Fig. 1) was also performed. The same FGFR1 variant was also found in her dizygotic twin sister and her mother, although her father had wild-type FGFR1 (Fig. 2). Considering that the majority of the pathogenic variants of the FGFR1 gene reported as IHH are missense variations, the variant of this report could be classified as 'likely pathogenic' according to the American Medical College of Medical Genetics and Genomics guidelines. Our patient was ultimately diagnosed with nIHH. Hence, she started sex hormone replacement therapy, and experienced pubertal changes in breast development as well as a growth spurt after 6 months on estrogen therapy. She eventually started calcium and vitamin D supplementation to improve her BMD. She had menarche at the age of 17 years; her last recorded adult height on a recent follow-up visit at the age of 17 years was 157 cm (-0.7 SDS), while her body weight was 49 kg (-0.8 SDS) (Table 2).

Pedigree of the cases.
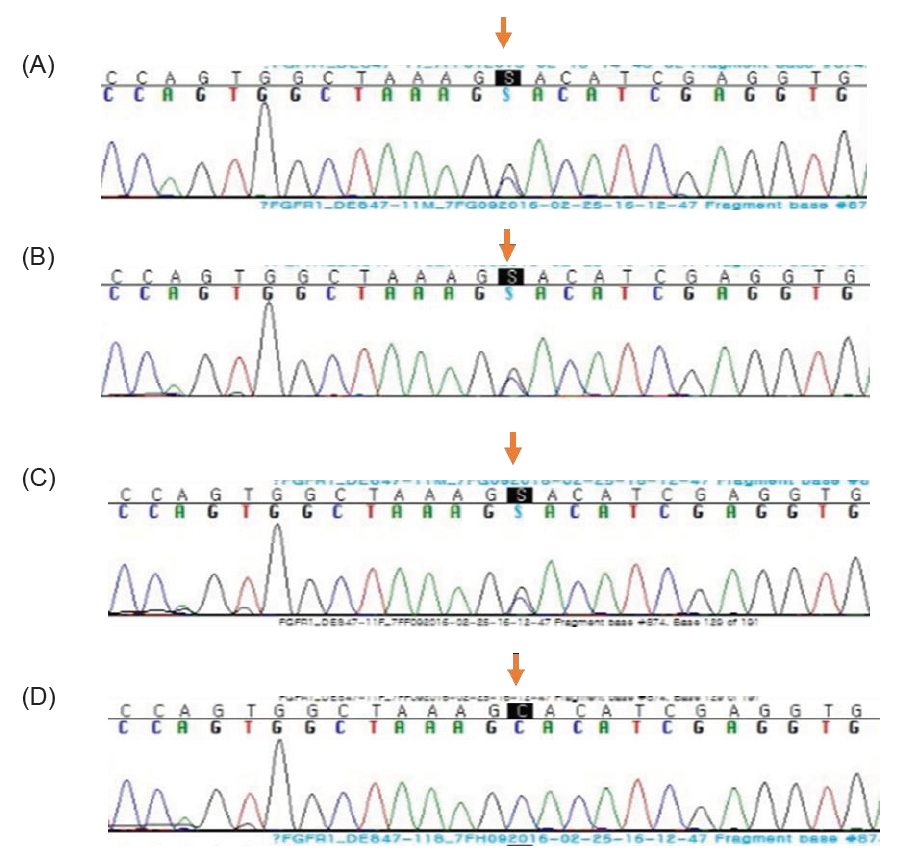
Exome sequencing for genetic analysis. (A) Heterozygous missense variant c.874C>G (p.His292Asp mutation) was found in the FGFR1 gene of patient 1 (the older sister of the dizygotic twin). (B) The same missense variant was found in the FGFR1 gene of patient 2 (the younger sister of the dizygotic twin). (C) The same missense variant was found in the FGFR1 gene of the mother. (D) Mutant sites in the FGFR1 gene of the father were validated to be wild-type.

Clinical findings of dizygotic twins with normosmic idiopathic hypogonadotropic hypogonadism after hormone replacement treatment
2. Case 2
The second sister was born at a birth weight of 2.0 kg (<3rd percentile). Unlike her twin and mother, she had no history of cleft palate. On physical examination at the age of 11.5 years, her height was 135.3 cm (-1.6 SDS) and her body weight was 31.4 kg (-1.1 SDS). Her breast and pubic hair development were Tanner stage I. No mirror movement, nystagmus, cerebellar ataxia, sensorineural hearing loss, or anosmia were detected. Her bone age was that of a 10-year-old girl. Spontaneous partial thelarche had occurred when she was 12.6 years old and her bone age was slightly accelerated, but she had no grow spurt in terms of height. Although her midparental target height was 163 cm, she started growth hormone treatment at the age of 12.6 years because of her mother's concern about the patient's short stature. It was discontinued after 1 year due to the parents'opinion that the response was not as effective as expected. Given that her pubertal development had not progressed by the age of 15 years and that her twin sister was diagnosed with nIHH via exome sequencing, we investigated her delayed puberty and short stature. Basal hormonal evaluation revealed low serum estradiol (<5.0 pg/mL) as well as suppressed levels of LH (0.2 IU/L) and FSH (4.4 IU/L). On GnRH stimulation, the peak LH and FSH levels were 6.0 IU/L and 5.2 IU/L, respectively (Table 1). Anterior pituitary function was otherwise normal according to the combined pituitary function test. Moreover, the KVSS test, sella MRI findings, and pelvic ultrasonography had normal findings like her sister. Her bone age was 13 years, indicating a 2-year delay relative to her chronological age. Her BMD (corrected for bone age) was reduced, with an L1–4 spine BMD z-score of -2.48. She was found to have the same FGFR1 variant c.874C>G (p.His292Asp) (Fig. 2) as had been discovered in her dizygotic twin sister and mother (Fig. 1), and was ultimately diagnosed with nIHH as well.
She started sex hormone replacement therapy immediately after her diagnosis, whereupon her pubertal development progressed with estrogen replacement, as did her height gain. On the latest follow-up visit at the age of 17 years, her pubic and axillary hair were both at Tanner stage III, while her breast development was Tanner stage IV. Her height was 157 cm (-0.7 SDS) and weight was 50 kg (-0.6 SDS). Menarche started at the age of 17 years (Table 2).
Discussion
Pulsatile secretion of GnRH into the hypophyseal portal circulation is essential for maintaining normal gonadotropin synthesis and secretion; the frequency of these pulses is a critical determinant of LH and FSH synthesis and secretion [11]. This specialized GnRH neuronal network plays a commanding role in the biologic hierarchy; it controls episodic gonadotropin secretion, modulates gonadal steroid feedback, and ultimately determines the initiation or suppression of pubertal development and fertility during the life cycle [12,13]. IHH is diagnosed if the lack of pubertal development is associated with reduced GnRH and gonadotropin release in the absence of an apparent cause.
We identified a novel heterozygous missense variant c.874C>G (p.His292Asp) in FGFR1 that was present in both twin sisters and their mother. IHH is caused by a large number of mutations in many different genes, and the mechanisms that underlie nIHH also involve mutations in many genes. Some of these genes encode proteins that regulate GnRH neuronal migration as well as its secretion or action. A number of these mutations have been identified only recently, and their roles in the regulation of reproduction are not yet well understood [1,3,14]. The FGFR1 gene is located on human chromosome 8 at position p11.23 (8p11.23), is composed of 24 exons, and encodes a tyrosine kinase receptor that mediates fibroblast growth factor signaling. FGFR1 encodes the FGFR1 receptor appear in olfactory and GnRH neuron development. Loss of FGFR1 function has been reported to elicit reproductive abnormalities ranging from severe autosomal dominant KS through fully penetrant nIHH to delayed puberty [15-17]. Its mutations were detected in 7% of 134 patients with nIHH, suggesting that this gene should be a key component of diagnostic panels for this condition [18]. As for the features of FGFR1 loss-of-function, cleft palate is found in up to 30% of patients, while cartilage abnormalities in either the ear or nose as well as some anomalies of the digits have been reported [19]. In our case, the older of the twins and their mother had cleft palate, while the younger twin did not, despite all having the same FGFR1 gene missense variants. Hence, it is notable that different clinical phenotypes may manifest in IHH patients that share the same FGFR1 gene variant [5]. Detailed phenotyping of individuals carrying identical FGFR1 mutations revealed a broad range of phenotypes, both within and between different kindreds; these range from apparent normality to isolated anosmia, pubertal delay, isolated cleft lip, IHH with reversal, nIHH, and even KS with craniofacial or skeletal defects [15]. This phenotypic variability may be a consequence of the inheritance of distinct mutations (i.e., digenic or oligogenic inheritance).
Most patients with IHH are diagnosed in late adolescence or early adulthood, as IHH is difficult to differentiate from other causes of delayed puberty [20]. Patients diagnosed in adulthood often present with infertility or even osteoporotic fractures. The delay in diagnosis might be caused by missed opportunities to diagnose IHH during adolescence, especially in female girls, since IHH has a male predominance of 3–5 to 1 [5,6]. In contrast to male patients, female counterparts exhibit no specific clinical signs of IHH, especially during the neonatal and childhood periods.
Currently, effective therapies are available for both male and female patients diagnosed with IHH that promote the development of secondary sexual characteristics and induce fertility. The prognosis for infertile patients has improved, as increasing numbers of patients with nIHH are now able to have children through medically assisted procreation. Hormonal replacement can induce puberty, as was the case in our patients. While estrogen administration increases uterine size, and combined estrogen and progestin treatment induces monthly withdrawal bleeding but not ovulation, fertility requires gonadotropin or GnRH therapy, each of which is effective [20]. Thus, effective treatment is available not only for inducing estrogenization, but also for successful development of fertility. Regardless of the therapy considered, patients and their families should be clearly informed that treatment is likely to be lifelong and will require regular monitoring for optimal benefit. With appropriate and long-term treatment, many of the long-term effects of hypogonadism can be minimized [20].
In addition to reproductive issues, patients with IHH are known to have bone health problems owing to the lack of sex steroids, which leads to osteoporosis. Although sex steroid treatment typically improves bone density, it does not fully reverse the phenotype [20]. In our patients, osteoporosis was revealed by densitometry even though they were still young. They both commenced vitamin D replacement immediately after their diagnosis. Notably, the IHH guideline recommends performing a bone density measurement in all patients at baseline as well as after 2 years of treatment and at the time of achieving their final height [20].
In conclusion, we report the first patients in Korea with familial nIHH caused by a missense variant in FGFR1. This finding provides new information regarding the features of nIHH as well as an additional reference for future precision medicine and prenatal diagnostic techniques. It is difficult to diagnose IHH based only on the constitutional delay of growth and puberty; therefore, physicians should be cognizant of this condition and carefully evaluate patients suspected of having it. Because effective treatment is available, age-appropriate sex hormone replacement or fertility induction therapy should be started as soon as the diagnosis is made. IHH is a life-long condition that requires ongoing management. Long-term and well-controlled studies are necessary to investigate the benefits of different therapeutic options on the quality of life and on fertility.
Notes
Ethical statement
This study was approved by the Institutional Review Board of the Ilsan Paik Hospital (ISPAIK 2019-07-038). We obtained written informed consent from the patients and their mother in order to report these findings.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
The authors would like to thank Dr. Ja-Hyun Jang (Green Cross Genome, Yongin, Korea) for his help with genetic analysis.