Persistent goiter with congenital hypothyroidism due to mutation in DUOXA2 gene
Article information

Abstract
Thyroid hormones are crucial for development of the central nervous system. Congenital hypothyroidism (CH) is the most common preventable disease resulting in mental retardation. A neonatal screening test (NST) can detect a mild form of CH that can be treated at an early age. Generally after 3 years of age, when most of the brain has matured, clinicians consider reevaluation of thyroid function for CH patients that have been identified with a normal thyroid gland at a normal position. This report presents three CH patients that developed normally, with persistent goiter despite thyroid hormone supplements. The patients’ initial thyroid-stimulating hormone (TSH) level after NST was 47, 157, and 57 mIU/L, respectively. Levothyroxine administration began at 1 or 2 months of age and was terminated after reevaluation at the age of 3, 15, and 5 years, respectively. However, 1 or 2 years later, they all resumed their medication due to increased TSH level coupled with newly developed or enlarged goiter. They all showed dual oxidase maturation factor 2 (DUOXA2) gene mutation: a homozygous mutation with DUOXA2 (c.413dupA; p.Tyr138*) in case 1, a presumed compound heterozygotic mutation with DUOXA2 (p.Tyr138*/p.Tyr246*) in case 2, and heterozygous mutations with DUOXA2 (c.738C>G; p.Tyr246*) and TPO (c.2268dupT; p.Glu757*) in case 3. When goiter persists or is newly developed despite a maintained euthyroid status, for those with transient CH history, follow-up to assess the thyroid function is recommended for at least 1 or 2 years, and genetic testing would be helpful. This study presents the first clinical cases of DUOXA2 mutation in Korea.
Introduction
Thyroid hormones affect the metabolism of all cells in the body and play a very important role in growth and neurodevelopment, particularly in children. Recently, the incidence of congenital hypothyroidism (CH) has increased to 1 in 1,000–2,000 live births worldwide, which has been confirmed by neonatal screening test (NST) [1]. CH is the most common disease that can be detected early, and appropriate treatment can prevent intellectual disability. Treatment is usually simple and inexpensive, mostly through prescription of thyroid hormone. Through early diagnosis and proper treatment, patients with CH can achieve normal height and intelligence.
Thyroid dysgenesis makes up about 75%–80% of etiology in CH cases, while dyshor-monogenesis accounts for approximately 15%–20% [2-4]. Thyroid dysgenesis includes athyrosis, ectopic thyroid, and hypoplasia. Thyroid agenesis or ectopic thyroid usually requires permanent thyroid hormone supplement. Except for athyrosis, general treatment for CH is to maintain levothyroxine until 3 years of age, when most brain development is complete. Reevaluation of thyroid function at several weeks after drug cessation is helpful for deciding whether to continue with treatment. Due to NST, mild cases of CH can be detected and treated. If the thyroid appears to be in a normal position, mild CH patients can terminate treatment, which is meant for transient types of CH, usually considered as dyshormonogenesis. Numerous studies have searched for a predictive variable between permanent and transient CH [5,6]. However, to date, no definite factor has been identified to differentiate permanent from transient forms of CH.
There are 7 steps in thyroid hormone biosynthesis: (1) transport of thyroid iodide, (2) thyroglobulin (Tg) synthesis, (3) oxidation and organification, (4) H2O2 generation, (5) coupling of iodotyrosyl residues of Tg, (6) endocytosis of colloid and hormone (thyroxine [T4], triiodothyronine [T3], diiodotyrosine, monoiodotyrosine) release, and (7) recycling of iodide [4]. The following genes are related to thyroid hormone synthesis: SLC5A5 (NIS), SLC26A4 (PDS), TG, TPO, DUOX2, DUOXA2, and IYD [4,7]. Goiter usually appears in CH when there is a defect in at least one of those genes. Determination of the genetic mutation causing CH assists in predicting whether the treatment will be permanent or transient. Park et al. [8] reported that DUOX2 mutations are commonly detected in CH patients in Korea. They also identified oxidase maturation factor 2 (DUOXA2) mutation, although they only counted the number and did not describe the phenotype.
We report 3 cases of DUOXA2 mutation in CH. This is the first clinical case report of DUOXA2 mutations in Korea.
Case reports
1. Case 1
A 4.5-year-old boy was referred to our clinic. He had received levothyroxine treatment for CH since 2 months of age. On his first visit to our clinic, he presented a huge goiter (7 cm×4 cm) (Fig. 1A) with elevated Tg (107.39 μg/L; reference range, 0–35 μg/L), although his thyroid function test (TFT) was normal (free T4 [fT4], 1.71 ng/dL; TSH, 3.24 mIU/L). His height was 109 cm (75th–90th percentile), body weight was 18.3 kg (50th–75th percentile), and all developmental milestones were normal.
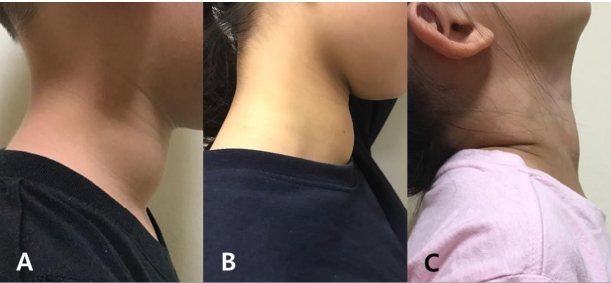
(A) Case 1 developed a new goiter 1 year after stopping levothyroxine treatment. (B) Case 2 had a enlarged goiter 2 years after discontinuing levothyroxine. She still has the goiter despite a euthyroid state with taking levothyroxine. (C) Case 3 developed a new goiter 2 years after treatment cessation. All 3 patients resumed treatment and still have a goiter, while maintaining a euthyroid status.
The patient was born at 38+5 weeks of gestation, with a birth weight of 3.48 kg. The initial neonatal screening TSH level was 47.14 mIU/L (reference range, <10 mIU/L), while the serum TSH level decreased to 6.28 mIU/L when he was 1 month old. One month later, not only had TSH level increased to 38.62 mIU/L, but goiter had developed, for which he was consequently started on levothyroxine. After commencement of treatment, his goiter was not palpable. When he reached 3 years of age, he was able to stop medication, as TFT results after 6 weeks of holding medication indicated normal ranges (fT4, 1.78 ng/dL; TSH, 5.26 mIU/L) with a palpable goiter. He resumed the medication at the age of 4 due to increased TSH level up to 24.34 mIU/L and because the goiter had become visible (Table 1).
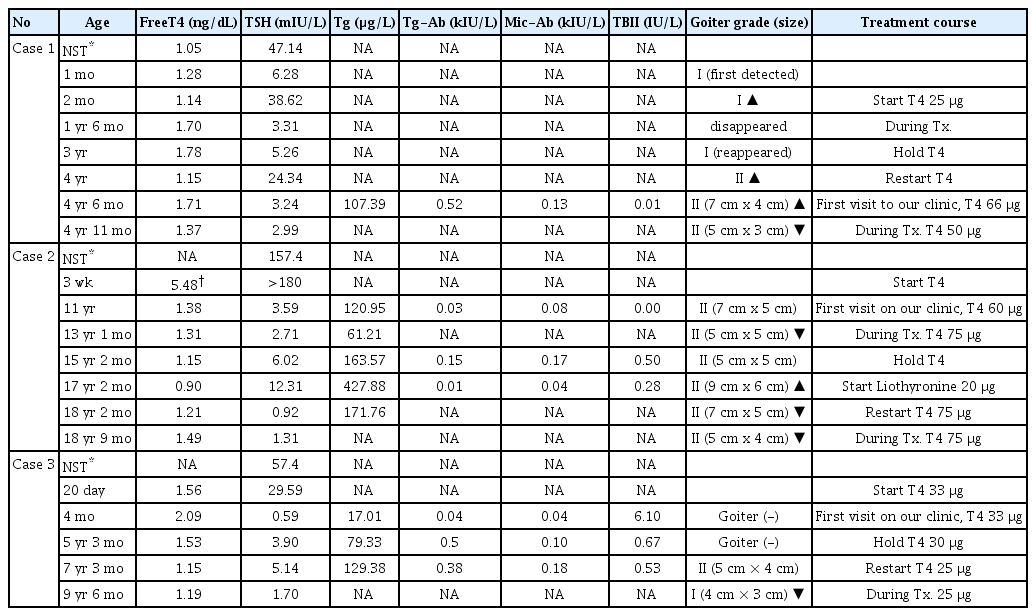
Clinical course and laboratory findings in the 3 cases
We performed next-generation sequencing (NGS) for hypothyroidism that covers 23 related genes (TSHR, PAX8, TSHB, NKX2-5, THRA, GNAS, FOXE1, NKX2-1, SLC5A5, TPO, TG, DUOXA2, DUOX2, SLC26A4, IYD, SLC16A2, THRB, POU1F1, PROP1, LHX3, TSHB, HESX1, TRH, TRHR) using Miseq (Illumina, San Diego, CA, USA) with 192X depth of coverage and greater than 99% accuracy. A homozygous mutation of DUOXA2 with c.413dupA (p.Tyr138*) was identified (Fig. 2A). He was an only child, and his parents refused to undergo genetic analysis. After 11 months of treatment, his goiter had slightly decreased (5 cm×3 cm) with normal TFT.
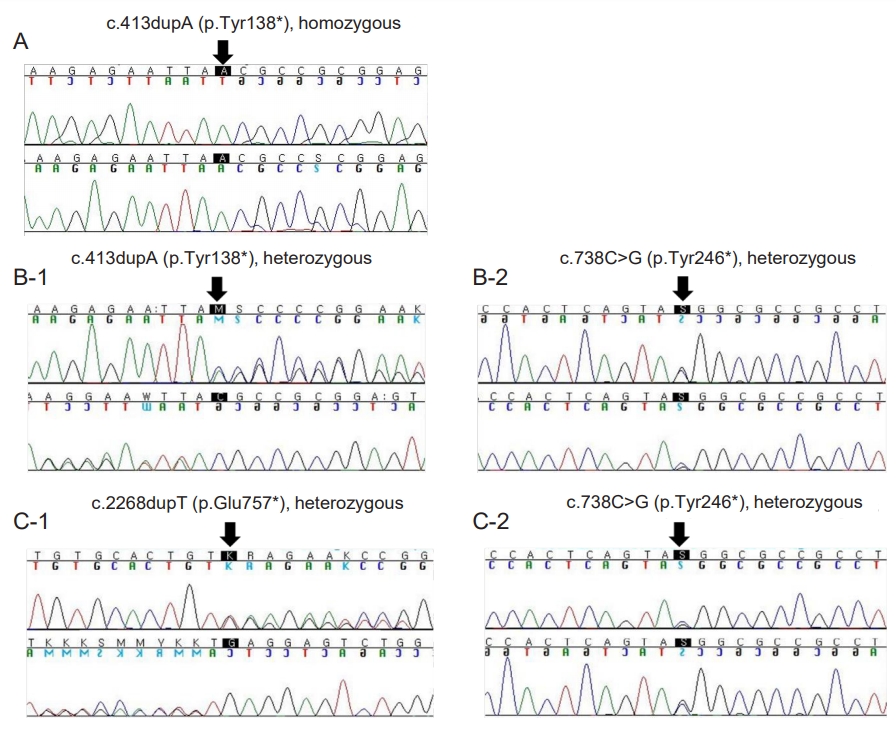
Next-generation sequencing for hypothyroidism revealed genetic mutations. (A) A homozygous mutation with DUOXA2 at c.413dupA (p.Tyr138*) was identified in case 1. (B) A presumed compound heterozygous mutation with DUOXA2 at c.413dupA (p.Tyr138*) (B-1) and c.738C>G (p.Tyr426*) (B-2) was identified in case 2. The mutation was assumed to be compound heterozygous based on her clinical presentation, although there was no confirmation as her parents refused to undergo genetic analysis. (C) Heterozygous mutations of both TPO at c.2268dupT (p.Glu757*) (C-1) and DUOXA2 at c.738C>G (p.Tyr246*) (C-2) were detected in case 3.
2. Case 2
An 11-year-old girl diagnosed with CH was referred to our hospital for a huge goiter (7 cm×5 cm) under levothyroxine medication. She was born at a gestational age of 39+0 weeks and body weight of 3.0 kg. The initial NST for TSH was 157.4 mIU/L and 225.6 mIU/L at 7 and 12 days old, respectively. She had started levothyroxine treatment at 3 weeks (total T4, 5.48 μg/dL [reference range, 4.5–12.5 μg/dL]; TSH >180 mIU/L). Her father, grandfather, and 3 uncles from her father's side had thyroid disease, though the exact diagnosis was unknown. The initial laboratory findings in our clinic were fT4 1.38 ng/dL; TSH 3.59 mIU/L; and Tg 120.95 μg/L. She had a height of 144.5 cm (50th–75th percentile), and a body weight of 42 kg (50th–75th percentile). She had normal intelligence and experienced menarche at the age of 12 with normal puberty progression. During persistent treatment with levothyroxine 75 μg, goiter size decreased to 5 cm×5 cm, and Tg level decreased (61.21 μg/L) compared to the initial level.
She was able to stop the treatment at 15 years of age, as her fT4 level was normal with mild elevation of TSH (6.02 mIU/L) after 6 weeks of no medication. We assessed a normal TSH response of thyrotropin releasing hormone stimulation test. A 99mTc scan revealed increased uptake of 18.9% (reference range, 1.7%–4.0%). After 2 years, liothyronine 20-μg medication was restarted as she complained of tiredness with a repeated huge goiter (9 cm×6 cm) (fT4, 0.90 ng/dL; TSH, 12.31 mIU/L; Tg, 427.88 μg/L) (Table 1) (Fig. 1B). All thyroid antibodies were normal. The goiter decreased slightly (7 cm×5 cm) after treatment restarted.
An NGS test for hypothyroidism was performed, and two nonsense mutations of DUOXA2, c.413dupA (p.Tyr138*) and c.738C>G (p.Tyr246*), were identified (Fig. 2B). Genetic analysis was recommended to her parents to confirm whether her mutations were compound heterozygous. Although her parents refused to perform the test, there were apparent clinical interpretation matched her genetic mutation to be regarded as compound heterozygous state. After identifying the genetic cause of her CH and goiter, we changed her medication from liothyronine to levothyroxine.
Under treatment, goiter size decreased to 5 cm×4 cm, although it was still big. Currently, core needle biopsy is being planned for 2 newly detected hypoechoic nodules (0.92 cm and 0.62 cm).
3. Case 3
A 4-month-old girl visited our clinic for CH treatment (fT4, 2.09 ng/dL; TSH, 0.59 mIU/L; Tg, 17.01 μg/L). She had started levothyroxine 20 days after birth (fT4, 1.56 ng/dL; TSH, 29.59 mIU/L). Her initial TSH level of NST was 57.4 mIU/L.
She was able to stop treatment at 5 years of age, as the TFT level was normal after 6 weeks without therapy (fT4, 1.53 ng/dL; TSH, 3.90 mIU/L; Tg, 79.33 μg/L). A 99mTc scan showed increased uptake of 6.9% (reference range, 1.7%–4.0%). At that point, goiter was neither visible nor palpable.
Two years later, however, a goiter (5 cm×4 cm) was detected by her mother. Her TFT result indicated subclinical hypothyroidism (fT4, 1.15 ng/dL; TSH, 5.14 mIU/L), and treatment with 25-μg levothyroxine was restarted (Table 1). An NGS test for hypothyroidism revealed heterozygous mutations for both DUOXA2 [c.738C>G (p.Tyr246*)] and TPO [c.2268dupT (p.Glu757*)] (Fig. 2C). The patient had a younger brother who was treated for transient CH until 18 months of age. Her parents declined genetic testing for him and for themselves. The patient still has a goiter (4 cm×3 cm) and is under 25-μg levothyroxine medication with an euthyroid status (Fig. 1C).
Discussion
The 3 cases of CH presented in this study with DUOXA2 mutations showed newly developed or enlarged goiter after terminating levothyroxine treatment. They all had to resume treatment due to increased TSH level within 1–2 years of treatment cessation.
Both oxidation of iodide and iodination of Tg by thyroid peroxidase are important steps for synthesizing thyroid hormone. Hydrogen peroxidase (H2O2), produced at the apical membrane of thyroid follicular cells, is a critical substrate for both steps. The dual oxidases (DUOX1 and DUOX2) play important roles for catalyzing H2O2 formation [9,10] The DUOXA2 gene, which is comprised of 6 exons and encodes a 320-amino acid transmembrane protein, is essential for DUOX2 maturation and activation. A mutation in DUOXA2 was first discovered in 2008 in 11 CH patients with partial iodide organification defects without TPO or DUOX2 mutation [11]. Mutation of DUOXA2 affects partial or complete iodide organification that can lead to dyshormonogenesis [11-13]. The phenotype associated with DUOXA2 mutation is usually inherited in an autosomal recessive manner, although some reports suggest autosomal dominant inheritance [13]. Regardless of inheritance, goiter is a characteristic clinical presentation. Because thyroid hormone production capacity is impaired, elevated TSH stimulates the thyroid follicular cells to compensate for defective function. This stimulation increases the Tg level as the thyroid enlarges to become a goiter. However, there is currently no clear explanation for why the goiter persists even with an euthyroid status in DUOXA2 mutation.
Case 1 featured the previously reported nonsense homozygous DUOXA2 (p.Tyr138*) mutation, a loss-of-function mutation. There was a report on Japanese siblings who had the same mutation but with a different phenotype [14]. The younger sister started levothyroxine treatment after she was diagnosed with CH at 13 days old with a high serum Tg level (>1,000 μg/L; reference range, 3.8–56.3 μg/L) and a goiter confirmed through ultrasonography (thyroid size, +5.9 standard deviation [SD] [15]). She had to continue medication, because her TSH level elevated after stopping medication at 3 years of age. In comparison, her elder brother had a normal neonatal screening TSH result and a normal TFT at 10 years of age. Based on elevated Tg (106 μg/L; reference range, < 30 μg/L) and a slightly enlarged goiter observed on ultrasonography (thyroid size, +1.7 SD [15]), mild defect of thyroid hormone producing capacity was suspected. Similarly, case 1 had a normal TFT period of 1 year without medication, the same as in the elder brother. However, case 1 had to resume treatment due to increased TSH with enlarged goiter, which was similar to the younger sister’s case. Therefore, based on these reports, there could be additional unknown factors regulating the expression of enzymatic activity.
Case 2 is regarded as a compound heterozygote DUOXA2 (p.Tyr138*/p.Tyr246*) mutation, although the parents were not genetically tested. The same mutation was first described in 2013 from China [16]. The mutation was detected in only 1 of 47 CH patients, and that patient was diagnosed with CH after NST, without a report of goiter. Recently, the same mutation was reported in a goitrous hypothyroidism fetus treated with intrauterine levothyroxine injection [17]. The goiter size in case 2 was the largest of our 3 reported cases, with the highest Tg level. To date, no reports have presented a correlation between thyroid nodule or cancer and DUOXA2 mutation. We are currently waiting for results for biopsies of the hypoechoic nodules.
Case 3 was identified to have each of the known pathogenic heterozygote mutations in DUOXA2 (p.Tyr246*) and TPO (p.Glu757*). She started levothyroxine 33-μg treatment at 20 days old and was on 25 μg at 10 years old, the smallest amount of the three cases. A mild form of CH coupled with goiter suggests mild defect in organification in this case. Since both TPO and DUOXA2 genes are inherited in an autosomal recessive manner, the identified mutations did not entirely explain the clinical aspects. Therefore, we considered large deletion/duplication or deep intronic mutation not indicated by the NGS test. Nonsense mutation p.Tyr246* is the first known pathogenic DUOXA2 mutation, found in a Chinese female with homozygotic state [11]. In that study, only heterozygotic mutation probands did not have TFT abnormality. In 2015, another Chinese female was reported to have homozygotic state with goiter and elevated Tg level [13].
Because there are 2 DUOXs and 2 DUOXAs in the thyroid gland, transient improvement of thyroid function may be explained by a "backup" of DUOX1/DUOXA1 due to lower RNA expression levels compared to DUOX2/DUOXA2 [18,19]. DUOX2 mutations are frequently associated with CH in Korea and Japan, and the prognosis is usually transient [2,8,20]. In contrast, DUOXA2 mutations are uncommon and seem to be more pathogenic compared to DUOX2 mutations. Since 2008, when DUOXA2 was first detected [11], there have been 10 reported patients, most from China, Japan, or Korea (Table 2). Goiter appears to be consistent with lengthy treatment. However, a definite genotype-phenotype correlation is limited for the DUOXA2 mutation. Therefore, additional case studies and longterm follow-up are required.

Summary of heterogenous genotype-phenotype correlation in DUOXA2 mutation
All three cases required resumption of levothyroxine treatment 1–2 years after treatment cessation. In conclusion, regular follow-up for TFT and physical examination of enlarged or newly developed goiter are recommended for at least 1 or 2 years after patients who terminate transient CH treatment. Additionally, genetic analysis is helpful to determine the cause of goiter.
Notes
Ethical statement
This study was approved by the Institutional Review Board of Soonchunhyang University Seoul Hospital (approval number: 2019-03-021). The parents of all cases offered informed written consent for genetic tests and using the results and photographs obtained from the patients.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This report was financially supported by Soonchunhyang University.