Identification of a novel variant in the PHEX gene using targeted gene panel sequencing in a 24-month-old boy with hypophosphatemic rickets
Article information

Abstract
Familial hypophosphatemic rickets (FHR) is a disorder characterized by phosphate wasting and hypophosphatemia due to defects in renal phosphate transport regulation. There are 4 known inherited forms of FHR that differ in their molecular causes. Very few studies have been conducted that focused on the molecular analysis of FHR in Koreans. Eighteen mutations of the PHEX gene have been identified to this date in Korea. Herein, we report the clinical case of a 24-month-old boy presenting with bowed legs and short stature. The biochemical profile showed hypophosphatemia with decreased tubular reabsorption of phosphate. Several family members were identified with short stature and genu varum. Therefore, he was diagnosed with FHR. To identify the molecular causes of FHR, we performed targeted gene panel sequencing and found a novel hemizygous missense variant, c.1949T>C (p.Leu650Pro), in the PHEX gene. This variant was also detected in the boy’s mother who exhibited genu varum and short stature.
Introduction
Familial hypophosphatemic rickets (FHR) is a disorder characterized by phosphate wasting and hypophosphatemia caused by defects in renal phosphate transport [1]. Four inherited forms of FHR have been reported so far based on their molecular origin [2-5]. The most common form is X-linked dominant hypophosphatemic rickets (XLHR; MIM 307800), which is caused by a loss-of function mutation of the PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome) gene [2]. Autosomal dominant hypophosphatemic rickets (MIM 193100) is caused by mutations of the FGF23 gene [3]. The autosomal recessive forms include: (1) autosomal recessive hypophosphatemic rickets 1 (MIM 241520) caused by mutations of the DMP1 gene [4]; and (2) autosomal recessive hypophosphatemic rickets 2 (MIM 613312) caused by mutations of the ENPP1 gene [5].
Characteristic clinical features of FHR include decreased serum phosphate levels and renal phosphate reabsorption, elevated alkaline phosphatase, lower than normal levels of 1,25-dihydroxyvitamin D, and normal serum levels of calcium and parathyroid hormones [5,6]. Clinical symptoms of FHR include deformity of the lower limbs, short stature, bone pain, and tooth abscess [1].
Very few studies until now have focused on the molecular analysis of Korean FHR patients, and the number of reports on hypophosphatemic rickets caused by the PHEX gene mutation in Korea is small [7-12].
We performed targeted gene panel sequencing of the PHEX, FGF23, DMP1, and ENPP1 genes in a 24-month-old boy with hypophosphatemic rickets and detected a hemizygous c.1949T>C (p.Leu650Pro) variant in the PHEX gene. The heterozygous form of the same variant was detected in the boy’s biological mother who presented with clinical symptoms similar to those of her son. Here, we report a novel hemizygous missense variant, c.1949T>C (p.Leu650Pro) of the PHEX gene.
Case report
A 24-month-old boy was brought to the Department of Orthopedics due to bowed legs. Radiographic findings showed bowed legs with fraying and widening of the distal femoral metaphysis (Fig. 1). The patient was suspected to have rickets and was referred to the Department of Pediatric Endocrinology. He was born by cesarean section due to premature rupture of membranes at 35 weeks and 4 days gestation. His birth weight was 2,720 g (5th‒10th percentile) and his birth height was 48 cm (10th‒25th percentile). He was the first child of nonconsanguineous Korean parents. At the time of presentation of the clinical symptoms, his height and body weight were 79.5 cm (-2.49 standard deviation score [SDS]) and 11.6 kg (-0.41 SDS), respectively. Developmental parameters for assessing the social, speech, and gross motor and fine motor functions as determined by the Korean developmental screening test were appropriate for his age. The measured heights of the father and mother were 165 and 141 cm, respectively. The mother had previously undergone surgical correction for genu varum. On further inquiry into the family history of the disease, we learned that several of her family members had also been diagnosed with short stature and genu varum (Fig. 2), with some of them having undergone surgical correction for genu varum.
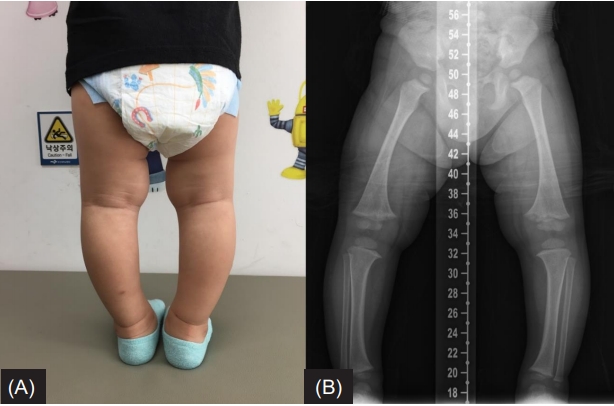
(A) The patient’s photograph taken at the age of 2 years shows bowed legs. (B) A radiograph of the lower legs reveals diffuse bone contour changes as well as fraying and widening of the distal femoral metaphysis with varus deformity.

Pedigree of the proband is consistent with a dominant mode of inheritance. The black symbols represent the affected individuals with short stature and genu varum. The asterisks (*) indicate the individuals who had undergone surgical correction for genu varum.
Physical examination revealed frontal bossing and bowing of the legs (Fig. 1A). Radiological findings displayed diffuse (nonfocal radiolucency) changes in bone contour as well as fraying and widening of the distal femoral metaphysis with varus deformity (quadriceps angle, 7°) (Fig. 1B). Laboratory tests revealed hypophosphatemia (2.9 mg/dL; normal range, 4.3‒5.4 mg/dL), normal calcium level (10.2 mg/dL; normal range, 8.5‒10.3 mg/dL), and increased alkaline phosphatase level (527 IU/L; normal range, 156–369 IU/L). The intact parathyroid hormone level was 18.50 pg/mL (normal range, 10–65 pg/mL), 25-hydroxyvitamin D3 level was 53.52 ng/mL (normal range, 9.7‒52.8 ng/mL), and 1,25-dihydroxyvitamin D3 level was 59.58 pg/mL (normal range, 19.6–54.3 pg/mL). Although the patient had hypophosphatemia, tubular reabsorption of phosphate (TRP) measured in a single-void urine sample was reduced to 78.2% (normal range, over 90%), and TRP measured in a 24-hour urine sample was also reduced to 79.0%. Based on the above results, the patient was suspected of having hypophosphatemic rickets.
We performed targeted gene panel sequencing to detect mutations in genes associated with hypophosphatemic rickets. Genomic DNA was extracted from the peripheral blood and the library was prepared using the TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA, USA), which enriches a 12-Mb region spanning 4,813 genes with clinical relevance. Massively parallel sequencing was performed on the Illumina NextSeq platform. Sequence reads were mapped to UCSC hg19 standard base for comparative analysis. The average depth of the panel was 96X, and percentage of bases above 10X was 98.3%. Genes related to FHR were PHEX, FGF23, DMP1, and ENPP1, and the percentages of bases above 10X for the exons and intronic flanking regions (20 bp) of these genes were 99%, 99%, 100%, and 97%, respectively. The results of targeted gene panel sequencing revealed a hemizygous c.1949T>C (p.Leu650Pro) variant in the PHEX gene, which was confirmed by Sanger sequencing using custom-designed primers (Fig. 3). There were no pathogenic variants in other genes. The c.1949T>C (p.Leu650Pro) variant in the PHEX gene was also found in his mother with short stature and genu varum (Fig. 3). In addition, this variant has been suggested to affect protein function in validation studies using in silico analyses (SIFT score, 0.001; PolyPhen-2(HVAR) score, 0.885; MutationTaster score, 1) [13-15].
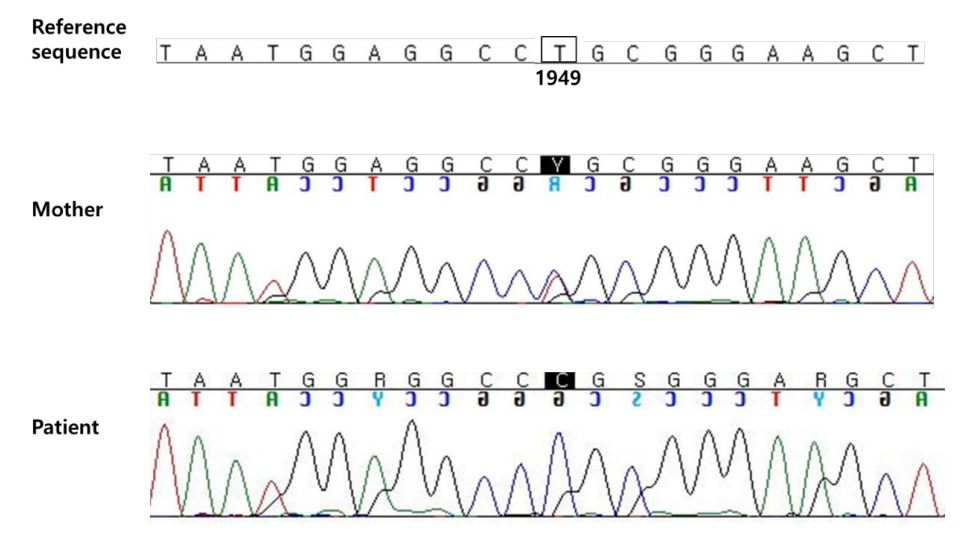
Partial genomic DNA sequence of the patient and patient’s mother shows the c.1949T>C (p.Leu650Pro) variant in the PHEX gene.
Therefore, the c.1949T>C (p.Leu650Pro) variant in the PHEX gene can be classified as a likely pathogenic variant according to the guidelines of the American College of Medical Genetics and Genomics.
The patient was put on an initial treatment regimen with alfacalcidol 0.5 μg/day (43 ng/kg/day) and elemental phosphorus 45 mg/kg/day in 4 divided dosages, and the alkaline phosphatase level returned to normal (363 IU/L) after 3 months of treatment. A year from the treatment start date, the patient’s height was measured to be 88 cm (-2.03 SDS), and the weight was 13.3 kg (-0.53 SDS). The degree of bowing of the legs did not increase. The patient is currently 3 years and 10 months old, is being treated with alfacalcidol 1.5 μg/day (95 ng/kg/day) and elemental phosphorus 50 mg/kg/day in 4 divided dosages, and has not shown any side effects associated with the medications (i.e., hypercalcemia, hypercalciuria, and hyperparathyroidism).
Discussion
In the present study, we report a hemizygous missense variant, c.1949T>C (p.Leu650Pro), in the PHEX gene in a hypophosphatemic rickets patient. To the best of our knowledge, this variant is novel and has not been reported in the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk,access date: March 29, 2019).
Since the PHEX mutation associated XLHR was first reported [2], 457 mutations have been reported by the HGMD with various mutation types from missense, nonsense, splicing site mutations, insertions, to deletions. A total of 18 different PHEX gene mutations have been reported in 6 articles in Korea [7-12]. The correlation between the PHEX gene mutations and phenotypes is unclear, and no genotype-phenotype correlation has been observed in Korean patients with hypophosphatemic rickets [8,16]. Recently Yang et al. [12] reported the first mosaic mutation in the PHEX gene in a Korean patient and summarized all the PHEX gene mutations reported in Korea.
A loss-of-function mutation of the PHEX gene causes XLHR, and XLHR is the most common form of FHR, accounting for 80% of all cases of FHR [1,2]. The PHEX gene, consisting of 22 exons is located at Xp22.1. The PHEX gene, a member of the M13 family of membrane-bound metalloproteases, encodes a 749 amino acid protein [2]. The c.1949T>C (p.Leu650Pro) variant is located at exon 19 and corresponds to the zinc-binding motif of the extracellular domain of the PHEX gene [17]. The wild type of the Leu650 residue is well-conserved from birds to mammals and humans, suggesting that amino acid changes are likely to affect protein function (source: http://www.h-invitational.jp/evola). We reviewed the c.1949T>C (p.Leu650Pro) variant for its frequency in various databases including 1,000 Genomes (2,504 whole-genome sequences, http://phase3browser.1000genomes.org), Exome variant server (6,503 exome sequences, http://evs.gs.washington.edu/EVS/), ExAC (60,706 exome sequences, http://exac.broadinstitute.org/), and gnomAD (125,748 exome sequences and 15,708 whole-genome sequences, https://gnomad.broadinstitute.org/) and they report no minor allele frequency.
The inactivating mutation of the PHEX gene, as well as mutations of the FGF23, DMP1, and ENPP1 genes increase the serum levels of fibroblast growth factor 23 (FGF23) [5,6,18]. FGF23 is a protein synthesized by osteoblasts and osteocytes, and it inhibits phosphate reabsorption in the renal tubules. Increased FGF23 levels lead to renal phosphate wasting, and consequently, hypophosphatemia [18]. Chronic hypophosphatemia causes bony deformity and short stature within a year after birth if left untreated.
The current treatment regimen consists of oral phosphate supplementation with multiple daily intakes to compensate for renal phosphate wasting and active vitamin D analogs (alfacalcidol or calcitriol) to compensate for 1,25-dihydroxyvitamin D deficiency [19]. Specific oral phosphate and calcitriol dose ranges administered at different life stages are as follows: infancy: oral phosphate 55-70 mg/kg/day and calcitriol 0.75-1.0 μg/day (alfacalcidol 1.5-2.0 μg/day); childhood: oral phosphate 45-60 mg/kg/day and calcitriol 0.5‐1.0 μg/day (alfacalcidol 1.0‐2.0 μg/day); and puberty: oral phosphate 35-50 mg/kg/day and calcitriol 0.75-1.5 μg/ day (alfacalcidol 1.5-3.0 μg/day). Daily doses of phosphate supplementation and vitamin D analogs are adjusted according to alkaline phosphatase levels, leg bowing, growth velocity, and parathyroid hormone level. Since vitamin D analogs can lead to side effects such as hypercalciuria and nephrocalcinosis, it is recommended that the urinary calcium/creatinine ratio should be measured every 3‐6 months and that renal ultrasound should be performed every year [19]. Even after appropriate pharmacotherapy, complete alleviation of bone deformities and restoration of growth cannot be achieved, and surgical correction of leg bowing may be needed in some cases [7,10].
Recently, a phase 2 trial using a human monoclonal antibody against FGF23 (burosumab) has been reported [20]. In this open label study, children aged 1‐4 years with XLHR from 3 hospitals in the USA received burosumab (0.8 mg/kg) via subcutaneous injection every 2 weeks for 64 weeks. Burosumab had a favorable safety profile, increased serum phosphorus, and improved rickets. In the future, these findings could alter the treatment modalities in patients with XLHR.
In conclusion, we identified a novel hemizygous missense variant, c.1949T>C (p.Leu650Pro), in the PHEX gene in a 24-month-old boy with FHR. Because several genes are related to FHR, targeted gene panel sequencing can be a useful molecular diagnostic tool.
Notes
Ethical statement
This study was approved by the Institutional Review Board of Pusan National University Hospital, Busan, Korea (approval number: 1807-018-069). Authors have received written informed consent from the parents of the child to use the data and the clinical picture for publication purposes.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by clinical research grant from Pusan National University Hospital in 2019 (2019-96).