A case of vitamin D hydroxylation-deficient rickets type 1A caused by 2 novel pathogenic variants in CYP27B1 gene
Article information

Abstract
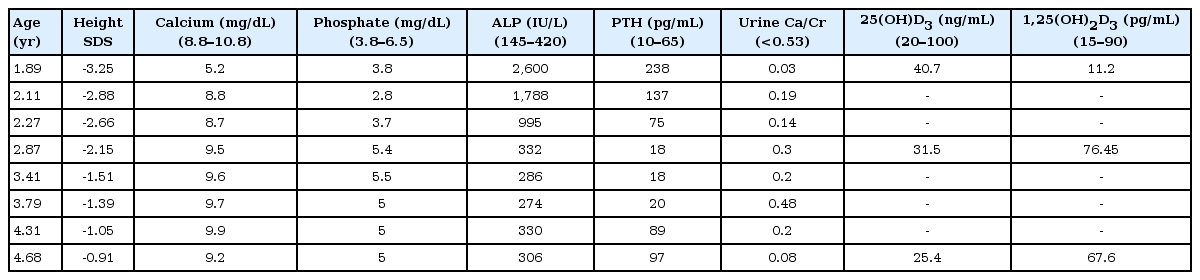
Vitamin D hydroxylation-deficient rickets type 1A (VDDR1A, OMIM 264700) is a rare autosomal recessive inherited disorder. Pathogenic variants in the CYP27B1 gene lead to loss of 1α-hydroxylase activity. We report the case of a 22-month-old toddler who presented with growth retardation and delayed development. The patient exhibited the typical laboratory findings of VDDR1A, including hypocalcemia (calcium: 5.2 mg/dL), elevated serum level of alkaline phosphatase (2,600 U/L), elevated serum level of intact-parathyroid hormone (238 pg/mL), low 1,25(OH)2D3 level (11.2 pg/mL), and normal 25(OH)D3 level (40.7 ng/mL). His height and weight were 76.5 cm and 9.5 kg, respectively (both <3rd percentile). The Bayley Scales of Infant and Toddler Development II indicated significantly delayed development (mental development index <50, psychomotor development index <50). The patient was a compound heterozygous for two novel pathogenic variants in the CYP27B1 gene: c.57_69del (p.Glu20Profs*2) and c.171dupG (p.Leu58Alafs*275), inherited from his mother and father, respectively. The patient showed remarkable improvement after treatment with calcitriol and calcium carbonate.
Introduction
Rickets is a systemic disease that affects bone growth due to disruption in calcium, phosphate, and/or vitamin D metabolism and defects in the mineralization of the osteoid matrix [1]. Genetic rickets disorders are rare and include disorders of vitamin D biosynthesis or its receptor activity that result in vitamin D deficiency or resistance. All these disorders present symptoms similar to those of rickets [2].
VDDR1A (OMIM 264700), also known as pseudovitamin D-deficiency rickets, is an autosomal recessive inherited disorder caused by pathogenic variants in the CYP27B1 gene, which encodes 25-hydroxy vitamin D-1-α-hydroxylase [3-6]. Treatment with physiologic replacement doses of 1α(OH)vitamin D3 or 1,25-dihydroxyvitamin D3 improves the clinical, biochemical, and radiological abnormalities [2]. Patients with VDDR1A are clinically normal at birth and are generally medically diagnosed at 1–2 years of age due to muscle weakness and poor growth and gross motor development. Some infants may develop hypocalcemic seizures or experience bone pain, which manifests as irritability [2].
Worldwide, rickets is most frequently caused by nutritional vitamin D deficiency, the peak age for which is the first year after birth when the child is breastfeeding [7]. Some patients with VDDR1A may be misdiagnosed with nutritional vitamin D-deficiency rickets, as they are clinically similar.
We report the case of a 22-month-old male who presented with delayed development and poor growth. The patient exhibited the typical laboratory findings of VDDR1A, including hypocalcemia, hypophosphatemia, elevated alkaline phosphatase (ALP) level, and elevated intact parathyroid hormone (PTH) level with low 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) level in the presence of normal 25-hydroxyvitamin D3 (25(OH)D3) level.
Case report
A 22-month-old male presented with poor growth, inability to walk, and delayed verbal expression. He could say only one word other than "mom" and "dad." He was born at the 40th week of gestation without perinatal problems to healthy non-consanguineous parents. At birth, his weight was 3.3 kg (-0.1 standard deviation score [SDS]) and his height was 50 cm (0.06 SDS). He was the second child of the family. The father's and mother's heights were 170 cm (-0.81 SDS) and 161 cm (-0.01 SDS), respectively. His elder sister (40 months old) exhibited normal growth and development. There was no family history of rickets.
After breast-feeding for 1 month, formula feeding had been initiated. At 5–6 months, weaning foods were started. At 15 months of age, he experienced frequent loose stools following milk feeding; therefore, feeding of milk was decreased. This resulted in aggravation of his dietary calcium deficiency. At 19 months of age, his mother started giving him vitamin D 400 IU daily.
His parents did not notice developmental delay until he was 12 months of age. However, he could not stand without support or walk by himself at 12 months of age. At that time, his parents did not worry about his delayed gross motor development, because his elder sister had started walking by herself at 14 months of age. According to his parents, after 12 months of age, he slept for longer durations, his intake of food and physical activity decreased, and he did not gain body weight. At 18 months of age, he had a fever and a subsequent generalized tonic-clonic seizure that lasted for a few minutes. This was regarded as a febrile seizure, and no medical evaluation was performed. He could not stand independently or walk until he was 22 months of age.
At 22 months of age, he was referred for evaluation of growth retardation and delayed development. On physical examination, classical features of rickets were revealed, such as frontal bossing, delay in the closure of the anterior fontanel, Harrison's groove, rachitic rosary, and palpable widening of the wrists and ankles. His height was 76.5 cm (<3rd percentile), his body weight was 9.5 kg (<3rd percentile), and his head circumference was 46.3 cm (10th–25th percentile).
Laboratory findings were as follows: hemoglobin 12.4 g/dL, white blood cell count 5,800/mm3, platelet count 286,000/mm3, aspartate transaminase 42 IU/L, alanine transaminase 17 IU/L, ALP 2,600 U/L, total cholesterol 132, protein 6.6 g/dL, albumin 5 g/dL, calcium 5.2 mg/dL, phosphate 3.8 mg/dL, ionized calcium 0.8 mmol/L, magnesium 1.7 mg/dL, blood urea nitrogen 11.1 mg/dL, creatinine 0.2 mg/dL, PTH 238 pg/mL, 25(OH)D3 40.7 ng/mL, 1,25(OH)2D3 11.2 pg/mL, pH 7.42, pCO2 33.9 mmHg, HCO3 21.5 mmol/L, and tubular reabsorption of phosphate 86.3% (Table 1). Echocardiogram showed QTc prolongation (QTc 0.47). Radiological evaluation showed fraying and cupping of the metaphyseal regions of the radius, ulnar, tibia, and fibula (Fig. 1). The Bayley Scales of Infant and Toddler Development II, at 22 months of chronological age, revealed significant global developmental delay: mental development index <50 and psychomotor development index <50 (reference: development index ≥85 is considered average). Cognition and language developmental profiles were equivalent to those of 12–14 months, and motor developmental profile was equivalent to that of 9–12 months.
Clinical and laboratory findings of the patient in relation to time
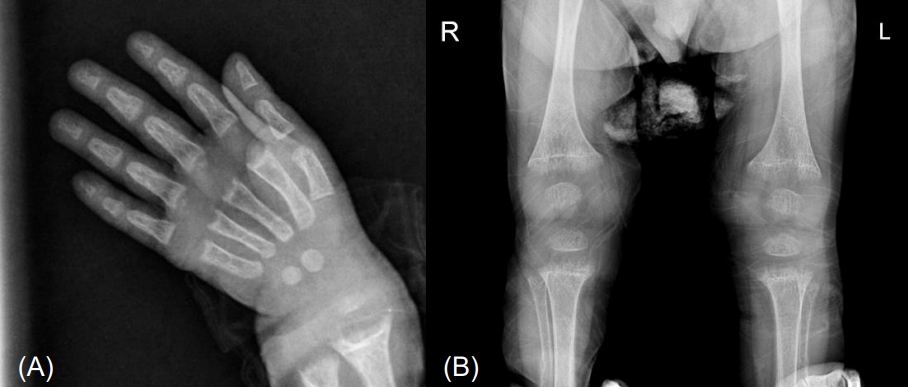
Radiological findings. Simple radiography shows fraying and cupping of the metaphyseal regions of the radius and ulnar (A), tibia and fibula (B). R, right; L, left.
CYP27B1 was analyzed by Sanger sequencing of the entire coding region, and 2 novel pathogenic variants (NM_000785.3: c.57_69del and c.171dupG) in exon 1 were found. The c.57_69del variant was predicted to substitute a glutamic acid with a proline at amino acid position 20 and a stop codon at amino acid position 22 (p.Glu20Profs*2). The c.171dupG variant was predicted to substitute a leucine with an alanine at amino acid position 58 and create a new reading frame (p.Leu58Alafs*275). These 2 variants were classified as pathogenic variants because both are very rare and are predicted to be null variants. In addition, family study revealed that c.57_69del was inherited from his mother, while c.171dupG was inherited from his father (Fig. 2). The elder sister was a heterozygous carrier of c.171dupG.

Genetic analysis of CYP27B1. (A) The sequencing chromatogram shows a heterozygous c.57_69del variant in the forward direction. The variant of the proband is inherited from the mother. (B) The chromatogram shows a heterozygous c.171dupG variant in the reverse direction. The variant of the proband is inherited from the father.
We initiated treatment with calcitriol 0.75 μg/day (80 ng/kg/day) with elemental calcium (60 mg/kg/day) for our patient. After 2–3 months of therapy, he was able to walk independently. However, there was persistent elevation in ALP (1,788 U/L) and PTH (137 pg/mL) levels. Therefore, we increased the dose of calcitriol to 1.0 μg/day. Over the next 12 months, the calcitriol dose was decreased to 0.5 μg/day because his dietary intake of calcium was sufficient, and calcium supplementation was withdrawn during this tapering process. During calcitriol (0.5 μg/day) therapy, high-normal serum levels of calcium (10.2 mg/dL) and lower-normal PTH values (14 pg/mL) were observed. We replaced calcitriol with alfacalcidol (Table 1). In the follow-up period, the physical and radiological abnormal findings had disappeared. No nephrocalcinosis was observed. He regained acceleration in growth. His most recent follow-up visit was at the age of 4 years 8 months; his height was 103.6 cm (10th–25th percentile) and weight was 16.2 kg (10th–25th percentile). He showed resolution of language delay. At 4 years 8 months of age, psychometric evaluation conducted according to the Korean Wechsler Preschool and Primary Scale of Intelligence showed a performance intelligence quotient (IQ) of 76, verbal IQ of 85, and full-scale IQ of 77, indicating below-average IQ and borderline intellectual functioning. Thus, the patient needs to be followed up with psychometric evaluation.
Discussion
Vitamin D requires 25-hydroxylation in the liver and subsequent 1a-hydroxylation in the kidneys for its activation of its nuclear vitamin D receptor and exertion of its biological activities. 1,25(OH)2D3 plays a central role in calcium homeostasis and bone metabolism [2]. The 25-hydroxyvitamin D 1-α-hydroxylase gene (CYP27B1) is located in chromosomal region 12q13.1-13.3, which is composed of 9 exons and is approximately 5 Kb in size [3-6]. Pathogenic variants of CYP27B1 result in compromised 1-α-hydroxylase activity. Affected patients have low serum levels of 1,25(OH)2D3 and normal or high levels of 25(OH)D3 [2].
Thus far, over 80 pathogenic variants spanning all exons of CYP27B1 have been reported in the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php).
Various types of pathogenic variants in CYP27B1 have been identified, mostly including missense and nonsense changes, along with splice site changes, insertions, deletions, and duplications. The 2 most frequently observed pathogenic variants are c.1166G>A (p.Arg389His), commonly found in French Canadian patients of the Quebec province owing to a founder effect [8], and c.1319_1325dup (p.Phe443Profs*24), which arose independently by de novo events in several populations [9]. In Korea, to our knowledge, 3 pathogenic variants, c.589+1G>A, c.1319_1325dup, and c.983G>A (p.Trp328*), have been identified thus far [10,11]. The c.589+1G>A variant is the most common pathogenic variant and is observed in 7 of 12 Korean alleles [10]. In the present study, two novel pathogenic variants (c.57_69del and c.171dupG) were identified, and both of them were null variants. They were absent or in low frequency in control databases, such as 1000 Genomes Project (http://phase3browser.1000genomes.org/index.html), gnomAD (http://gnomad.broadinstitute.org/), and Korean Reference Genome Database (http://152.99.75.168/KRGDB/menuPages/rstInfo.jsp). The minor allele frequencies of c.57_69del and c.171dupG were none and 0.000007959, respectively, in the gnomAD database. Family study confirmed these two variants located in trans.
While Durmaz et al. [12] reported a good genotype–phenotype correlation in VDDR1A, Tahir et al. [13] reported no association between variant type and disease severity but rather remarkable phenotypical diversity between families carrying identical mutations. Intake of calcium and vitamin D metabolites and some other factors may influence α-hydroxylase activity.
Differential diagnosis between VDDR1A and nutritional rickets can be made on the basis of a low or inappropriately normal 1,25(OH)2D3 level and unresponsiveness to vitamin D treatment [14]. Differentiation from hypophosphatemic rickets is required in some VDDR1A cases with normal serum calcium levels [12]. In VDDR1A, serum phosphate levels decrease with an increase in PTH level and renal phosphate excretion. However, in hypophosphatemic rickets, serum PTH levels do not increase. In the majority of VDDR1A cases, 1,25(OH)2D3 levels are low or undetectable; however, some cases have normal 1,25(OH)2D3 levels, leading to diagnostic confusion [15,16].
In a study by Wang et al. [15], 2 patients with normal 1,25(OH)2D3 levels were reported. One patient carried a homozygous variant (c.566A>G;p.Glu189Gly) and exhibited normal serum calcium, phosphate, and 1,25(OH)2D3 levels but high serum PTH levels. The other patient carried a compound heterozygous mutation (c.1027C>T;p.Leu343Phe and c.1166G>A;p.Arg389His) and exhibited normal serum 1,25(OH)2D3 levels but high serum PTH levels. Mutations in these patients induced partial enzyme activity in vitro. A milder phenotype is associated with pathogenic variants with residual enzyme activity (c.566A>G;p.Glu189Gly, c.1027C>T;p.Leu343Phe, and c.305G>A;p.Gly102Glu) [15,17]. VDDR1A should be considered in patients exhibiting a phenotype of rickets and showing normal or increased 25(OH)D3 levels although the 1,25(OH)2D3 levels are normal [16].
Despite the need for lifelong treatment, one patient had spontaneous cessation of calcitriol treatment [12]. The patient presented with seizures due to severe hypocalcemia at 4 months of age. She responded well to calcitriol; however, there was a spontaneous improvement at the age of 11 years. As her variant (c.1319_1325dup;p.Phe443Profs*24) alters the reading frame and creates a premature stop, resulting in complete loss of 1a-hydroxylase activity, this is probably due to 1a-hydroxylase activity exerted by a non-CYP27B1 enzyme [12].
There were good responses to treatment with calcitriol (10–400 ng/kg/day) or alfacalcidol (80–100 ng/kg/day) [2]. The dose depends upon the severity of disease and body weight. Treatment is usually initiated at higher doses, followed by lower maintenance doses [2]. Starting calcitriol in infancy results in the correction of abnormalities without serious adverse events [18]. Adequate intake of dietary calcium (30–75 mg/kg/day of elemental calcium) is recommended. Management should result in low-normal serum levels of calcium (8.5–9 mg/dL), normal phosphate levels, and high-normal PTH levels [2,14]. Monitoring of 24-hour urinary calcium excretion (remain <4 mg/kg/day) and the ratio of excretion of urinary calcium to creatinine in spot urine (remain <0.8 [at ≤6 months], <0.6 [at 7–12 months], <0.53 [at 1–3 years], <0.39 [3–5 years], <0.28 [5–7 years], and <0.21 [>7 years]) is recommended [19].
To conclude, we report a case of VDDR1A presenting in a 22-month-old toddler with growth retardation and delayed development, in whom 2 novel pathogenic variants were identified. Although VDDR1A is a rare disorder, some milder cases may be missed; therefore, VDDR1A may be more prevalent than reported [15]. Genetic analysis is important for making an accurate diagnosis.
Notes
Ethical statement: This report was approved by the Institutional Review Board (IRB) of the Daegu Catholic University Medical Center, Daegu, Korea (IRB No. CR-18-178-L). The need for informed consent was waived by the IRB.
Conflict of interest: No potential conflict of interest relevant to this article was reported.