A novel CHD7 mutation in an adolescent presenting with growth and pubertal delay
Article information

Abstract
Mutations in the CHD7 gene, encoding for the chromodomain helicase DNA-binding protein 7, are found in approximately 60% of individuals with CHARGE syndrome (coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear abnormalities and/or hearing loss). Herein, we present a clinical case of a 14-year-old male presenting for evaluation of poor growth and pubertal delay highlighting the diagnostic challenges of CHARGE syndrome. The patient was born full term and underwent surgery at 5 days of life for bilateral choanal atresia. Developmental milestones were normally achieved. At age 14 his height and weight were -2.04 and -1.74 standard deviation score respectively. He had anosmia as well as prepubertal testes and micropenis (4 cm×1 cm). The biological profile showed low basal serum testosterone and gonadotropins (testosterone, 0.2 nmol/L; luteinizing hormone, 0.5 U/L; follicle-stimulating hormone, 1.3 U/L), and otherwise normal pituitary function and normal imaging of the hypothalamic-pituitary area. The constellation of choanal atresia, anosmia, mild dysmorphic features, micropenis and delayed puberty were suggestive of CHARGE syndrome. Targeted genetic testing of CHD7 was performed revealing a de novo heterozygous CHD7 mutation (c.4234T>G [p.Tyr1412Asp]). Further paraclinical investigations confirmed CHARGE syndrome. Despite the presence of suggestive features, CHARGE syndrome remained undiagnosed in this patient until adolescence. Genetic testing helps clarify the phenotypic and genotypic spectrum to facilitate diagnosis, thus promoting optimal follow-up, treatment, and appropriate genetic counselling.
Introduction
CHARGE syndrome (MIM 214800) is a rare genetic disorder characterized by a constellation of several congenital anomalies (coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear abnormalities and/or hearing loss). It was first described independently by Hall and Hittner in children with a combination of the hallmark congenital malformations [1]. The acronym and initial diagnostic criteria for CHARGE Syndrome were introduced by Pagon in 1981 [1,2]. Currently, clinical diagnosis is based on the combined Blake (1998, revised in 2006) [1] and Verloes' (2005) diagnostic criteria [3].
It occurs in approximately 1:10,000 live births and incidence varies from one in 8,500 to 15,000 depending on geographic region and clinical practices [2]. Patients with CHARGE syndrome exhibit a broad and variable clinical presentation [4]. The molecular basis of CHARGE syndrome was first described in 2004 by Vissers and colleagues. Mutations in CHD7 encoding of the chromodomain helicase DNA-binding protein 7, an ATP-dependent chromatin remodeler have been demonstrated to be the major genetic cause of CHARGE syndrome, underlying approximately 60% of cases [4]. The mutational spectrum is predominantly characterized by heterozygous single-nucleotide variants affecting protein function (44% nonsense, 34% frameshift, 11% splice site, and 8% missense) [5]. Whole-exon deletions or microdeletions of 8q12.1 (the chromosomal band including CHD7), are found in less than 5% of cases [5] Notably, almost all CHARGE syndrome patients are sporadic cases, yet a few familial cases have been documented [6]. No clear genotype-phenotype correlation exists [7].
We present the diagnostic challenges of CHARGE syndrome using the clinical case of a male adolescent presenting with delayed puberty in which genetic testing revealed a novel causative mutation in CHD7.
Case report
A 14-year-old Caucasian male presented for endocrine evaluation of poor growth and pubertal delay. He was the youngest of three children born to a nonconsanguineous couple. The family history was notable for delayed puberty in both parents. The mother reported having menarche at age of 15 while the father indicated he developed later than his peers having experienced his growth spurt at approximately age of 16. The mother received fertility treatment for her first pregnancy, yet subsequently conceived twice spontaneously with uneventful pregnancies and deliveries. The parents were otherwise healthy, with normal height (father: +1.08 standard deviation score [SDS], mother: +0.21 SDS) and no dysmorphic features. The patient was born at 41 weeks gestation (birth weight 3,350 g (-0.13 SDS); length 50 cm (-0.05 SDS); Apgar scores: 8/8/8). Bilateral choanal atresia was suspected clinically in the first days of life and mixed (fibrotic and osseous) choanal atresia (predominantly fibrotic) was confirmed via computed tomography (CT) imaging (Fig. 1). Endoscopic laser surgery was performed at 5 days of life with serial surgical interventions at 1 month and 3.5 months. Panendoscopy did not reveal any additional anomalies and ultrasound revealed normal urinary tract structures without visceral malformation. He achieved global developmental milestones within expected timeframes yet he did not begin walking until 18 months. Interestingly, the parents retrospectively reported that he often appeared to struggle with balance during infancy. At 3.8 years he underwent unilateral surgical repair of an inguinal hernia. His medical history was otherwise unremarkable. He reported a reduced sense of smell that had never been formally investigated. Pubarche was noted at age of 13. Charting his growth did not reveal any deflection as both height and weight parameters were between -1.5 and -2 SDS since the age of 5.

Axial computed tomography scan at day 3 of life at the reference level of the pterygoid plates, showing bilateral choanal atresia (arrows).
In the first visit in our clinic at 13.5 years, his height (Holter Stadiometer) was 139.9 cm (-2.04 SDS) and weight was 30.0 kg (-1.74 SDS). He exhibited normal segmental proportions (arm span: 138 cm, upper versus lower segment: 0 SDS). Clinical evaluation revealed a left low-set ear with helix anteversion and an ogival palate (with presence of a dental apparatus). The patient was unvirilized with Tanner A1 P2 G1 with prepubertal testes (2 mL bilaterally by Prader orchidometer) and micropenis (4 cm×1 cm; reference range, 5.5–12.8 cm [8]).
At 14 years, growth velocity was slow at 3.1 cm/yr (-2 SDS) and bone age was delayed by 2.5 years compared to chronological age, consistent with his lack of pubertal development. Formal smell testing ("Burghart Sniffin' Sticks Identification Test 16") confirmed anosmia (score 5/16, <5th percentile). Basal serum hormone profiling indicated prepubertal serum testosterone (0.2 nmol/L [0.1–1.0 {T1} via liquid chromatography mass spectroscopy, LC-MS/MS]) in the setting of low gonadotropins (LH: 0.5 U/L [0.5–2.5 {G1}]), FSH: 1.3 U/L (<3 [G1]) via the Cobas 6000 (Roche Diagnostics, Rotkreuz, Switzerland) using electrochemiluminescence immunoassay. Normal basal hormonal levels suggested intact thyrotrope, corticotrope and somatotrope axes (Table 1). Sertoli-cell function, as assessed by AMH (443.6 pmol/L [5–800 pmol/L]) and Inhibin B (86.4 pg/mL [35–182 pg/mL]), was normal for his prepubertal stage. There was no evidence of any chronic disease or functional causes for his hypogonadotrophic hypogonadism (Table 1).

Laboratory testing
The constellation of choanal atresia, anosmia, mild dysmorphic features, micropenis and delayed puberty were suggestive of CHARGE syndrome. After obtaining informed consent from the patient and his family, Sanger sequencing was performed on DNA extracted from peripheral blood. The patient was found to harbor a heterozygous CHD7 mutation (c.4234T>G [p.Tyr1412Asp]). This is a private variant, not seen in large control databases such as the 1,000 Genomes or ExAC. Structural modelling revealed that the Tyr residue at position 1412 is located in the core of the helicase C domain, and the change to an Asp is predicted to have a major detrimental effect on the structural stability of CHD7 as a large buried hydrophobic amino acid is replaced by a small charged hydrophilic one (Fig. 2). This variant is predicted to be deleterious by both sorting intolerant from tolerant-Algorithm and PolyPhen-2. The mutation was not detected in his parents, and is therefore a de novo mutation. Subsequent cranial magnetic resonance imaging (MRI) showed abnormal lateral and posterior semicircular canals (Fig. 3A) with vestibular and cochlear malformation (Fig. 3B). Additional findings included a verticalized sella turcica, a decreased hypophyseal volume (160 mm3) for his age (Fig. 3C), hypoplastic gyri, deformed left VII cranial nerve, right olfactive bulb aplasia and left olfactive bulb hypoplasia (Fig. 3D). Echocardiography did not reveal any cardiac malformation and echocardiogram was normal. The ophthalmologic investigation was within normal limits, notably without coloboma. Vestibular function assessment showed normal utricular (subjective vertical testing and ocular vestibular-evoked myogenic potential [VEMP] test) and saccular function (cervical-VEMP). The video head impulse test used to identify peripheral vestibular deficits revealed posterior and lateral semicircular canal dysfunctions on both sides, consistent with the anomalies found on the MRI. Audiometry was normal. Luteinizing hormone-releasing hormone stimulation testing (Table 1) triggered only minor increase in LH and FSH (8.3 mU/L and 4.4 mU/L, respectively, LH:FSH ratio 1.88). Bone densitometry via dual X-ray absorptiometry (Lunar iDXA-210430) showed normal bone density for his bone age (Lumbar spine z-score: -1.7 SDS) according to the International Society for Clinical Densitometry references. No neuropsychological evaluation was performed as our patient followed normal educational track without notable difficulty.
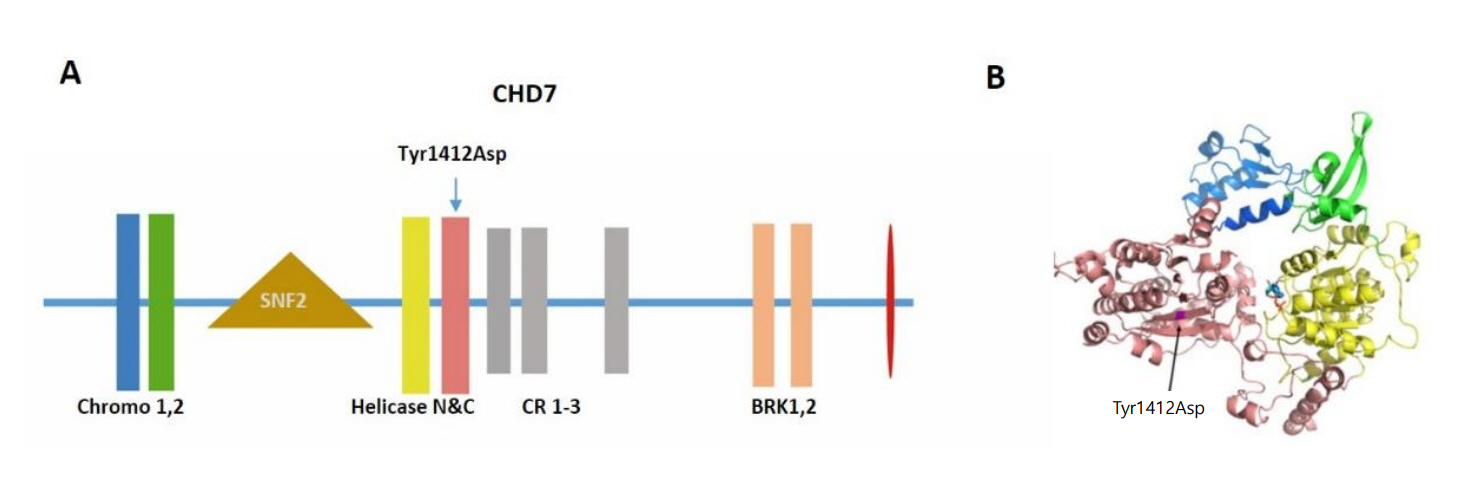
(A) The schematic representation of CHD7 protein and the location of mutation Tyr1412Asp. The functional domains are indicated in the schema. (B) The structural modeling of chromodomains and helicase domains of CHD7 and the location of Try1412Asp. Tyr residue at position 1412 is located in the core of the helicase C domain. The change to an Asp is predicted to have a major detrimental effect on the structural stability of CHD7 as a large buried hydrophobic amino acid is replaced by a small charged hydrophilic one.

(A-D) Imaging studies showing midline and intraauricular malformations. (A) Magnetic resonance imaging (MRI) (axial T1 with gadolinium) at 14 years of age, showing hypoplastic lateral and posterior semicircular canals (long arrows) and dysplastic vestibule (short arrow). (B) MRI at 14 years of age, showing Mondini dysplasia of the partition of the cochlea. (C) MRI at 14 years of age, showing decreased anterior pituitary volume (160 mm3). (D) MRI at 14 years of age, showing olfactory bulb aplasia (R) and hypoplasia (L). R, right; L, left.
Based on these findings and the Verloes' 2005 Criteria [3], the patient was diagnosed with CHARGE syndrome. Specifically, he exhibited 2 major criteria (choanal atresia and hypoplastic semicircular canals) and 2 minor criteria (hypothalamohypophyseal dysfunction/gonadotropin deficiency and abnormal external/middle ear) (Table 2). Low dose testosterone (50 mg via intramuscular injection monthly) was initiated at 14.6 years and improved growth velocity (from 3 to 9 cm/yr) and development of secondary sexual characteristics. No significant testicular enlargement was observed during this treatment and pubertal induction is ongoing.

Discussion
The combination of choanal atresia, anosmia, mild nonspecific dysmorphia, micropenis and pubertal delay in our patient suggested CHARGE syndrome. Genetic testing led to the identification of a de novo CHD7 mutation (p.Tyr1412Asp). Indeed, according to current genetic guidelines [9], several lines of evidence support that the identified CHD7 variant is pathogenic: (1) it is a private variant, i.e., not seen in the general population [10]; (2) it is located in a critical functional domain of CHD7 and is predicted to be deleterious by multiple computational tools; and (3) the de novo nature of this mutation is consistent with the absence of family history of CHARGElike features, while a heterozygous mutation is coherent with the autosomal dominant inheritance of CHARGE syndrome. This finding is concordant to other studies showing a high incidence of de novo changes in CHARGE syndrome [6]. The presence of a CHD7 gene mutation triggered further investigation that subsequently revealed additional clinical features and ultimately enabled the diagnosis of CHARGE syndrome.
During neonatal life, the patient’s choanal atresia had been considered an isolated congenital malformation, and thus further clinical and molecular investigations were not pursued. Consequently, CHARGE syndrome remained undiagnosed until adolescence. As with other syndromes with variable clinical presentation and several hallmark features, the initial diagnosis hinges on the awareness of the constellation of critical features, followed by careful clinical assessment. Choanal atresia is associated with other congenital malformations in approximately half of cases [11]. Further, a study by Burrow et al. [12] observed that CHARGE syndrome was diagnosed in 26% of patients with choanal atresia, and associated anomalies were more frequent in patients with bilateral choanal atresia. Similarly, the study by Kim et al. [13] found that CHARGE syndrome accounted for 4 of 27 cases (15%) of choanal atresia, yet was more frequent in patients with bilateral cases (4 of 14, 31%). Mutations in the CHD7 gene should therefore be tested in all newborns presenting with bilateral choanal atresia to facilitate early detection of CHARGE cases.
The vast phenotypic spectrum of CHARGE syndrome and the variable presentation in patients carrying presumed pathogenic CHD7 mutations have recently raised the debate of whether CHARGE syndrome should be considered a clinical diagnosis, a molecular diagnosis, or both [7,14]. In a recent study by Bergman and colleagues, 18 of 131 (14%) and 22 of 124 (17%) of patients harboring a mutation in CHD7 could not be clinically diagnosed with CHARGE syndrome based on the Blake’s and Verloes' diagnostic criteria, respectively. The authors proposed that CHD7 analysis should not be solely restricted to patients fulfilling current diagnostic criteria. The authors suggested that CHD7 analysis could be considered in the presence of any cardinal features, rather than limiting it to the 3 cardinal features; coloboma, choanal atresia, semicircular canal hypoplasia. Further, the presence of CHARGE syndrome features in a family member could also be a potentially suggestive feature [7]. Hale et al. [14] recently proposed new diagnostic criteria for CHARGE syndrome diagnosis by expanding the list of minor features and including CHD7 gene mutation as a major diagnostic criterion. Notably, 17% of patients with a CHD7 mutation do not meet current diagnostic criteria for CHARGE [6]. Accordingly, some have recommended CHD7 analysis in cases of suspected CHARGE syndrome, i.e., patients who do not meet current diagnostic criteria [6]. Considering the present case, a high clinical suspicion and thorough clinical evaluation combined with genetic testing enabled an accurate diagnosis. Notably, the strict adherence to the Hale criteria would have potentially led to earlier diagnosis in the present case. In our patient, neonatal diagnosis of CHARGE syndrome could have led to the evaluation of the central activation of the gonadotropic axis during minipuberty (2–3 months of life), and subsequently earlier intervention during adolescence in order to induce secondary sexual characteristic development concurrently with his peers.
The diagnosis of CHARGE syndrome may lead to an early treatment strategy of testosterone replacement aimed to restore a normal sex steroid milieu. This treatment might be switched to exogenous gonadotropin therapy or pulsatile GnRH therapy in case of continuous deficient secretion of gonadotropins or absence of reversal [15]. Pubertal delay and hypogonadotropic hypogonadism are common in patients with CHARGE syndrome. Indeed, hypogonadotropic hypogonadism occurs in nearly two-thirds of patients harboring CHD7 mutations [16]. Conversely, CHD7 mutations are found in 5%–8% of patients diagnosed with Kallmann syndrome (KS) [2,17,18]. Jongmans et al. [17] have proposed CHD7 sequencing in KS patients if one or more additional CHARGE syndrome features are present. Missense mutations are more prevalent in KS and are mostly transmitted through mildly affected fertile probands. Therefore, clinical and molecular assessment of family members may help expand the phenotypic and genotypic spectrum of CHARGE syndrome. Reversal of hypogonadic hypogonadism has been observed even in patients with a truncating mutation in the CHD7 [19].
Whether the patient has constitutional delayed of puberty or congenital hypogonadotropic hypogonadism, the treatment remains the same in adolescence presentation [20]. Due to the possibility of spontaneous activation of the gonadal axis, pubertal induction with low doses of testosterone seems reasonable. However, in cases of confirmed congenital hypogonadotropic hypogonadism, gonadotropin therapy is also a plausible treatment strategy and has the added benefit of inducing testicular growth and fertility potential [18].
In conclusion, we present a novel CHD7 mutation in a patient with a typical CHARGE syndrome (according to Verloes' diagnostic criteria) who remained undiagnosed until adolescence. The late detection was likely due to the lack of other signs/symptoms typically observed in CHARGE syndrome such as coloboma, hearing loss, and facial palsy.
Due to the benefits of prompt diagnosis and required followup of these patients, we recommend genetic testing in suspected cases which do not fulfill the complete clinical diagnostic criteria. Genetic testing promotes understanding of CHARGE syndrome development and clinical presentation, and may promote prompt diagnosis and thus timelier clinical care and access to genetic counselling.
Notes
Ethical statement: The study follows the ethical principles of the Declaration of Helsinki. The patient and his family gave their written consent to the publication of the case report.
Conflict of interest: No potential conflict of interest relevant to this article was reported.