Infantile hypercalcemia with novel compound heterozygous mutation in SLC34A1 encoding renal sodium-phosphate cotransporter 2a: a case report
Article information

Abstract
Idiopathic infantile hypercalcemia is characterized by hypercalcemia, dehydration, vomiting, and failure to thrive, and it is due to mutations in 24-hydroxylase (CYP24A1). Recently, mutations in sodium-phosphate cotransporter (SLC34A1) expressed in the kidney were discovered as an additional cause of idiopathic infantile hypercalcemia. This report describes a female infant admitted for evaluation of nephrocalcinosis. She presented with hypercalcemia, hypercalciuria, low intact parathyroid hormone level, and high 1,25-dihydroxyvitamin D3 level. Exome sequencing identified novel compound heterozygous mutations in SLC34A1 (c.1337G>A, c.1483C>T). The patient was treated with fluids for hydration, furosemide, a corticosteroid, and restriction of calcium/vitamin D intake. At the age of 7 months, the patient's calcium level was within the normal range, and hypercalciuria waxed and waned. Renal echogenicity improved on the follow-up ultrasonogram, and developmental delay was not noted. In cases of hypercalcemia with subsequent hypercalciuria, DNA analysis for SLC34A1 gene mutations and CYP24A1 gene mutations should be performed. Further studies are required to obtain long-term data on hypercalciuria and nephrocalcinosis.
Introduction
Idiopathic infantile hypercalcemia (IIH) is a rare disease entity of hypercalcemia characterized by failure to thrive, dehydration, vomiting, nephrocalcinosis, and hypercalcemia [1]. In the past, IIH was diagnosed after exclusion of other diseases related to development of hypercalcemia, such as Williams-Beuren syndrome, hyperparathyroidism, diuretic usage, and excess vitamin D intake [2]. Mutations in CYP24A1, which encodes 24-hydroxylase, responsible for degradation of 25-hydroxyvitamin D (25-OH-D) and 1,25-dihydroxyvitamin D (1,25-OH-D), were discovered as a cause of IIH [3]. However, a recent report described mutations in SLC34A1, which encodes the sodium-phosphate cotransporter, resulting in phosphate depletion and inappropriate production of 1,25-hydroxyvitamin D with subsequent hypercalcemia [4]. Herein, we describe an infant with IIH caused by a novel mutation in SLC34A1 and provide a literature review.
Case report
A 28-day-old Korean female presented with increased renal echogenicity that was detected on prenatal ultrasonography at 28 weeks gestation. She was the second child of unrelated parents. The mother of the neonate was healthy, and the father had a history of nephrolithiasis. The sister of the neonate was 6 years old and healthy. The patient was born at term after an uncomplicated pregnancy and normal delivery with a birth weight of 2.95 kg. The infant was exclusively fed breast milk. A vitamin D supplement was not given to the patient or her mother.
Poor feeding, hypotonia, lethargy, and irritability were not noted. Stool frequency was one time per week. At admission, the patient's height and weight were 52.4 cm (25th percentile) and 3.7 kg (15th–25th percentile), respectively. Urine output measured 7.7 mL/kg/day. On physical examination, no dysmorphic facial features of Williams-Beuren syndrome were found. Subcutaneous necrosis was not found. A renal ultrasonogram revealed bilateral medullary nephrocalcinosis (Fig. 1). A transthoracic echocardiogram showed 2 atrial septal defects without cardiovascular signs of Williams-Beuren syndrome.

(A) Medullary nephrocalcinosis of the left kidney on renal ultrasonography in the patient at admission. (B) Improved medullary nephrocalcinosis of the left kidney on the follow-up renal ultrasonography after 3 months.
Results of the laboratory investigations confirmed hypercalcemia and hypophosphatemia. Serum levels of total calcium and pH-adjusted ionized calcium were 12.8 mg/dL (normal range, 8.8–10.8 mg/dL) and 3.33 mEq/L (normal range, 2.2–2.5 mEq/L), respectively. The serum level of phosphate was 4.3 mg/dL (normal range for age, 4.8–8.2 mg/dL). The albumin level, which can affect the serum level of total calcium, was within the normal range (4.1 g/dL). Blood urea nitrogen and creatinine levels were 21 mg/dL and 0.35 mg/dL, respectively. Hypercalciuria was noted. The calcium/creatinine ratio of spot urine was 2.81 (normal for age, <0.8), and 24-hour urinary calcium excretion was 20.4 mg/kg (normal range, <4 mg/kg). Results of the blood gas analysis revealed no metabolic acidosis that could be related with renal tubular acidosis. The intact parathyroid hormone (PTH) was suppressed to 3.6 pg/mL (normal value, 15.0–65.0 pg/mL), and the PTH-related protein level was <1.1 pmol/L. The 25-OH-D level was 13.01 ng/mL (normal range, >20 ng/mL), and the 1,25-OH-D level was 71.27 pg/mL (normal range, 19.6-54.3 pg/mL). Serum levels of sodium, potassium, glucose, and magnesium and the complete blood cell count were within normal range.
Under the suspicion of infantile hypercalcemia, we conducted targeted exome sequencing. The process of targeted exome sequencing consisted of genomic DNA extraction, exome sequencing, filtering, analysis, and Sanger sequencing validation in the patient and her parents. Library preparation and massively parallel sequencing were conducted with the TruSight One sequencing panel (Illumina, San Diego, CA, USA) and NextSeq (Illumina), respectively. Generated reads were aligned to the hg19 human reference sequence. No mutation of CYP24A1 was identified. However, SLC34A1 missense mutations were identified in the compound heterozygous state in the patient (c.1337G>A, c.1483C>T) and were validated by Sanger sequencing (Fig. 2). Sanger sequencing of the parents revealed that c.1337G>A was inherited from the heterozygotic father, resulting in substitution of glycine with aspartic acid. Allele frequency of p.Gly446Asp was 0.01% in the Genome Aggregation Database and 0.05% in the Korean Reference Genome. The c.1483C>T was inherited from the heterozygotic mother, resulting in substitution of arginine with cysteine. Allele frequency of p.Arg495Cys was 0.02% in the Genome Aggregation Database and 0.05% in the Korean Reference Genome. These 2 variants had been interpreted as variants of unknown significance, and they have not been previously reported in IIH.

Sanger sequencing confirmation of compound heterozygous mutations of SLC34A1. (A) Heterozygous mutation of c.1337G > A in exon 12 of the SLC34A1 gene. (B) Heterozygous mutation of c.1483C > T in exon 13 of the SLC34A1 gene.
Although we did not perform an in vitro functional test, in silico analysis using the SIFT tool (http://sift.jcvi.org; PMID: 19561590) and PolyPhen-2 tool (http://genetics.bwh.harvard.edu/pph2/index.shtml; PMID: 20354512) suggested that the amino acid change was deleterious. Considering that IIH is an inherited autosomal recessive trait and the patient's elevated level of 1,25-OH-D, the compound heterozygous mutations inherited from each parent could be regarded as the patient’s cause of infantile hypercalcemia.
Clinical course of the patients is demonstrated in (Fig.3. To treat the patient's hypercalcemia, intravenous hydration with normal saline was started on the second day of hospitalization. Intravenous furosemide was also administered to the patient (1 mg/kg/day) at that time. After 7 days of hospitalization, the patient's serum calcium level decreased to 11 mg/dL, but ionized calcium was still elevated (3.26 mEq/L). For this reason, a corticosteroid (hydrocortisone, 3 mg/kg/day) was administered for 1 week. All medications were stopped at the age of 45 days. Thereafter, the patient was fed low-calcium, vitamin D-free formula (Calcilo XD, Abbott Nutrition, Columbus, OH, USA) for 3 months.
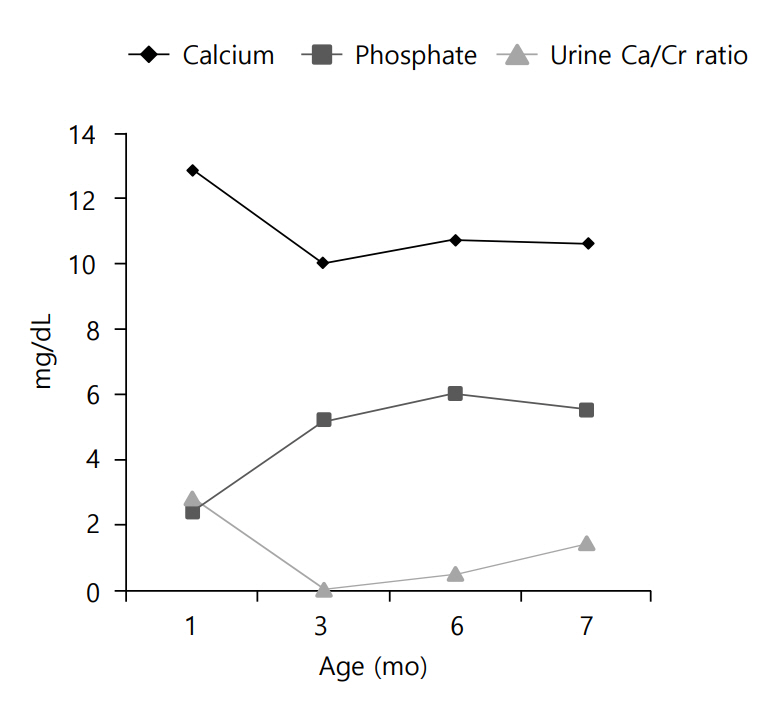
Changes of serum calcium, phosphate, and urinary calcium/creatinine (Ca/Cr) ratio.
Currently, at the age of 7 months, the patient's calcium and phosphate levels are within normal range, and hypercalciuria waxes and wanes. The previously noted renal echogenicity improved on the follow-up ultrasonogram, and developmental delay was not noted.
Limited data on natural history and long-term prognosis of IIH are available. Two siblings (1 boy and 1 girl) with mutations in SLC34A1 resulting in the substitution of histidine for arginine at position 495 were reported recently. The boy presented with hypercalcemia and nephrocalcinosis at 18 months [5].
Discussion
Calcium and phosphate are important components of bone mineralization and cellular activities, and the serum levels of calcium and phosphate are controlled within narrow limits by the kidney [6]. Schlingmann et al. [3] revealed that mutations in CYP24A1, which encodes 24-hydroxylase, the key enzyme for degradation of 25-OH-D and 1,25-OH-D, were causative for infantile hypercalcemia.
Recently, mutations in SLC34A1 encoding sodium-phosphate cotransporter 2a (NPT2a) were reported to lead to hypercalcemia, hypercalciuria, hypophosphatemia, and accumulation of 1,25-OH-D3. NPT2a and NPT2c (encoded by SLC34A3) are expressed in the kidney and reabsorbed as filtered phosphate [7]. Vitamin D, PTH, and fibroblast growth factor 23 (FGF23) are the major control mechanisms of calcium and phosphate. FGF23 is a phosphatonin that restricts renal reabsorption of phosphate through NPT2a. Additionally, FGF23 activates 24-hydroxylase and inhibits 1α-hydroxylase, resulting in decreased level of 1,25-OH-D [4].
Patients with mutations in SLC34A1 have decreased ability for renal phosphate reabsorption. The subsequent hypophosphatemia reduces the production of FGF23, resulting in activation of 1α-hydroxylase and inhibition of 24-hydroxylase. Consequently, patients present with increased level of 1,25-OH-D that induces hypercalcemia, hypercalciuria, and nephrocalcinosis [8].
Patients with infantile hypercalcemia present with vomiting, polyuria, dehydration, hypotonia, failure to thrive, and nephrocalcinosis [3]. One study noted that polyuria was induced by disturbance of urine concentration in rats [9]. Nephrocalcinosis was associated with hypercalciuria in another study [10]. The prevalence of nephrocalcinosis in patients with IIH due to mutation in NPT2a has not been identified. In the literature, however, all 18 patients with mutations in SLC34A1 had nephrocalcinosis [4,5,11]. Among patients with hypercalcemia and subsequent hypercalciuria, infantile hypercalcemia must be considered.
In the present case, the patient's father had a history of nephrolithiasis. Even a heterozygous mutation carrier presented with nephrolithiasis and renal phosphate leakage [4,12]. Therefore, patients who are asymptomatic heterozygous carriers should undergo additional evaluations.
Treatment of acute hypercalcemia includes rehydration and loop diuretics. Aggressive hydration with normal saline restores intravascular volume and enhances calcium excretion. Furosemide (0.5–1 mg/kg, every 6 hours) promotes calciuresis [13]. Other treatment options include a corticosteroid, calcitonin, and ketoconazole [14]. Ketoconazole inhibits 1α-hydroxylase at doses of 3–9 mg/kg [15]. Oral phosphate therapy at a dose of 0.5–1 mmol/kg/day has been documented in a report of a patient who developed hypercalcemia due to mutations in SLC34A1 [4]. Long-term management of hypercalcemia includes a low-calcium diet and avoidance of vitamin D. However, calcium and vitamin D restriction can be associated with diseases such as osteoporosis.
Long-term management of nephrocalcinosis includes high fluid intake, potassium citrate, and thiazide [16]. Urinary citrate binds to calcium and forms a soluble compound, resulting in reduction of calcium precipitation. Thiazide increases tubular calcium absorption and decreases renal calcium excretion. Although optimal dosing is not yet established, a daily dose of 0.5–2 mg/kg was effective in the decrement of urine calcium in children [17].
In conclusion, clinicians need to consider testing for mutations of SLC34A1 in patients with hypercalcemia, hypercalciuria, and nephrocalcinosis. Further studies in similar patients that explore the clinical course, long-term prognosis, and efficacy of a supplement with phosphate are required.
Notes
Ethical statement: Approval from the Keimyung University Dongsan Medical Center Institutional Review Board was waived for this case study. Informed consent was obtained from the patient's parents for publication.
Conflict of interest: No potential conflict of interest relevant to this article was reported.