De novo a novel variant of CaSR gene in a neonate with congenital hypoparathyroidism
Article information

Abstract
Autosomal-dominant hypocalcemia with hypercalciuria (ADHH) is a genetic disease characterized by hypoparathyroidism with hypercalciuria. Most patients with ADHH have calcium-sensing receptor (CaSR) gene mutations. The CaSR gene controls parathyroid secretions, and mutations in this gene can be detected via changes in serum calcium level. The activating mutation of the CaSR gene results in familial or sporadic ADHH. Most activating mutations of the CaSR gene are reportedly de novo missense mutations. This is the first case report of a novel activating variant of the CaSR gene in a neonate with congenital hypoparathyroidism with hypomagnesemia and hypercalciuria. We also report the 3-month follow-up management of the patient.
Introduction
Autosomal-dominant hypocalcemia with hypercalciuria (ADHH) is a genetic disease characterized by hypoparathyroidism with hypercalciuria [1]. Patients with ADHH show varying degrees of hypocalcemia. Although convulsions due to hypocalcemia can occur, most patients are asymptomatic or show intermittent limb numbness, muscular contractions, and laryngeal spasms [2]. Most patients with ADHH are known to have mutations of the calcium-sensing receptor (CaSR) gene [3]. Bartter syndrome, which is accompanied by hypocalcemia, hypomagnesemia, hypercalciuria, secondary hyperaldosteronism, and hypokalemia, can also develop due to activating mutation of CaSR gene [4]. CaSR is a G-protein-coupled receptor in cell membranes, which is predominantly located in the parathyroid gland and renal tubule; it detects minute changes in serum calcium levels and, consequently, controls parathyroid secretions. In addition, it participates in the renal regulation of positive ion concentrations through intracellular signaling pathways [5,6]. Generally, when the calcium concentration in the blood is lowered, the secretion of parathyroid hormone (PTH) increases, which then increases the reabsorption of calcium in the kidneys, ultimately leading to elevated blood calcium levels. As such, the parathyroid glands play a pivotal role in regulating systemic calcium homeostasis. Further, there is an inverse relationship between the secretion of PTH and extracellular calcium concentrations. When this homeostasis is disturbed, complications such as hypercalcemia and hypocalcemia can arise [7]. In patients with chronic hypocalcemia, CaSR actions increase the expression of the PTH gene and stimulate the proliferation of parathyroid cells, thereby maintaining the homeostasis of parathyroid functions. In contrast, in patients with hypercalcemia, the synthesis and secretion of PTH are suppressed.
Depending on the actions (inactivating or activating) of CaSR, hyper- or hypoparathyroidism can occur [1]. Hannan et al. [8] reported that 70 CaSR mutations had been confirmed at the time of their study, and 25 mutations associated with ADHH were reported. Activating mutations of CaSR gene, which are also referred to as heterozygous gain-of-function, result in familial or sporadic ADHH [9]. Most activating mutations of CaSR gene have been reported to be missense mutations. In 2004, Hu et al. [10] reported a de novo variant of CaSR gene in monozygotic twins. This study reports a novel activating variant of CaSR gene that occurred de novo in a Korean neonate who had congenital hypoparathyroidism with hypomagnesemia and hypercalciuria.
Case report
A 5-day-old female neonate was transferred to the neonatal intensive care unit (NICU) at the Kyungpook National University Children’s Hospital because of seizures. The patient was born through normal vaginal delivery at 39 weeks and weighed 3.2 kg. There was no notable finding during previously conducted prenatal care. Facial convulsions and muscular contractions in the limbs were observed on postnatal day 2. The patient was transferred to the NICU on postnatal day 5 because the symptoms persisted. Laboratory investigations revealed a serum calcium level of 4.7 mg/dL (normal range, 9.0–10.6 mg/dL), ionized calcium level of 2.2 mg/dL (normal range, 4.8–4.92 mg/dL), phosphate level of 13.2 mg/dL (normal range, 4.8–8.2 mg/dL), serum magnesium level of 1.29 mEq/L (normal range, 1.44–3.12 mEq/L), intact PTH (i-PTH) level of 2.6 pg/mL (normal range, 10–65 pg/mL), and 25-hydroxyvitamin D3 level of 13.95 ng/mL (normal range, 8.0–51.9 ng/mL) (Fig. 1).
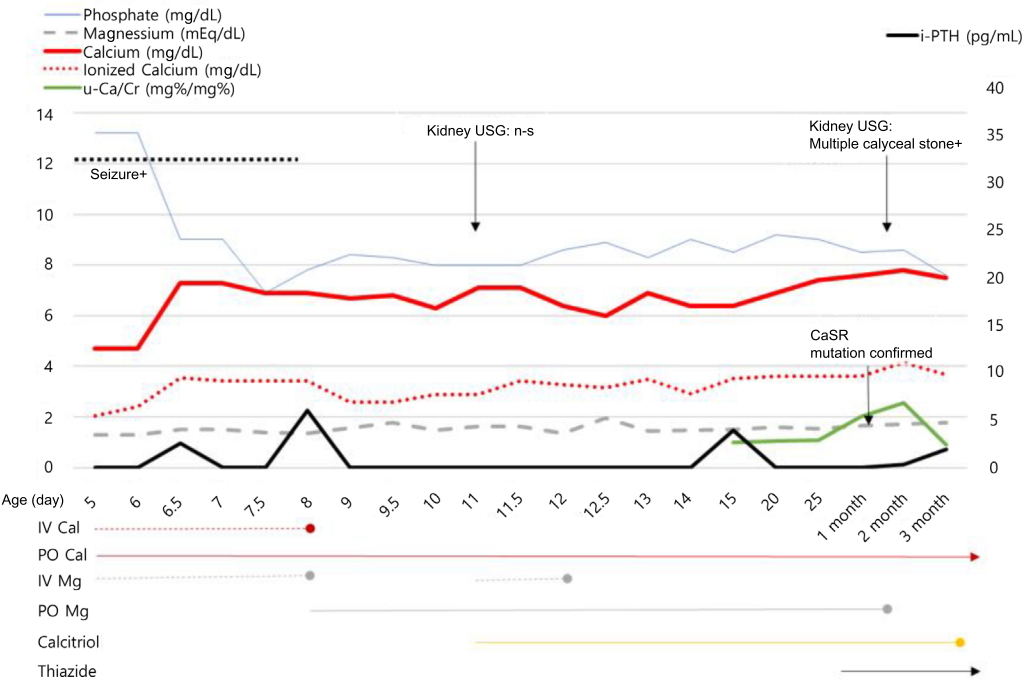
The serial changes in serum phosphate, magnesium, calcium, ionized calcium, i-PTH, and u-Ca/Cr ratio, and medication for 3 months, are shown. IV Cal, intravenous calcium; PO Cal, calcium carbonate per oral; IV Mg, intravenous magnesium; PO Mg, elemental magnesium per oral; i-PTH, intact parathyroid hormone; u-Ca/Cr, spot urine calcium/creatinine.
The morphology of the patient's face, limbs, and fingers did not show any abnormalities. An atrial septal defect of 3.4-mm size was noted on cardiac ultrasonography, and the results of renal ultrasonography were normal. Although the patient’s thymus glands were normal on chest ultrasonography, we tested for a 22q11.2 microdeletion because of the neonatal hypocalcemia. The result was negative. No abnormal finding was observed in auditory tests. The neonate's parents did not have a history of convulsions and muscular contractions, and nobody in her family had a history of kidney stones.
Because the patient presented with convulsions at the time of admission, calcium (10-mg/kg bolus) and magnesium (2.5 mg/kg/day) was also administered intravenously. And she was also administered elemental calcium (75 mg/kg) in the form of oral calcium carbonate. Until postnatal day 7, the convulsions continued intermittently and the hypocalcemia (serum calcium level, 6.9 mg/dL) and hypomagnesemia (serum magnesium level, 1.4 mEq/L) remained unresolved. Thus, calcium was administered continuously through intravenous injections and the magnesium dose was doubled to 5 mg/kg/day. The convulsions disappeared on postnatal day 8; therefore, the continuous intravenous calcium and magnesium administration was stopped. Because the hypocalcemia persisted (serum calcium level, 6.0–7.1 mg/dL), the dose of elemental calcium was increased to 180 mg/kg/day. Nevertheless, on postnatal day 11, the hypomagnesemia (serum magnesium level, 1.49 mEq/L) and hypocalcemia (serum calcium level, 6.4 mg/dL) did not improve. Intravenous magnesium injections (5 mg/kg/day) and calcitriol (0.25 μg/day) were added (Fig. 1). The patient was maintained on a low-phosphate diet formula because of hyperphosphatemia (phosphate level, 8.0–9.0 mg/dL). In blood tests repeated during the follow-up period, inappropriately low PTH levels (i-PTH, 0–6 pg/mL) were observed. Therefore, tests were conducted to screen for gene mutations associated with hypoparathyroidism, calcium abnormalities, and magnesium abnormalities (GCMB, GATA3, TBCE, CaSR, GNA11, preproPTH, and TRPM6 genes). The dose of calcitriol was increased gradually (from 0.25 μg/day to 1.5 μg/day). Because the spot urine calcium/urinary creatinine ratio (mg%: mg%) was elevated (1.0–2.02 [normal level, <0.8 for neonates]) at 1 month old, thiazide (0.5 mg/kg/day) was additionally administered to decrease urinary calcium excretion. The patient was subsequently followed up for 2 weeks in the NICU and discharged.
Targeted exome sequencing in the patient was performed using the TruSight One Sequencing Panel, resulting in a heterozygous novel variant c.2474A>T(p.Tyr825Phe) in exon 7 of CaSR gene confirmed by Sanger sequencing. No same variant was noted in the Sanger sequencing conducted for the parents. Thus, the patient was found to have a de novo novel activating vatiant of CaSR gene (Fig. 2). The in-silico analysis score of the variant yielded a SIFT score of 0 and a Polyphen-2 HVAR score of 0.994.

We identified a novel variant of calcium-sensing receptor (CaSR) gene in the patient and no variant in her parents. A heterozygous A to T transition is shown at position 2474 in exon 7 of CaSR gene, changing Tyr to Phe (T825P).
When the results were obtained, the patient had been discharged and was taking calcium carbonate (elemental calcium dose of 180 mg/kg/day), calcitriol (0.75 μg bid), magnesium (0.25 mEq/kg/day), and thiazide (0.5 mg/kg/day). The patient was rehospitalized to modify and manage the doses. At the time of readmission, the patient was 2 months old. In a blood test conducted upon readmission, the following measurements were obtained: serum calcium level of 8.0 mg/dL (normal range, 9.0–10.6 mg/dL), ionized calcium level of 4.2 mg/dL (normal range, 4.8–4.92 mg/dL), phosphate level of 8.1 mg/dL (normal range, 4.8–8.2 mg/dL), magnesium level of 2.2 mEq/L (normal range, 1.92–3.12 mEq/L, at age 7 days–2 years), i-PTH level of 0.34 pg/mL (normal range, 10–65 pg/mL), and 25-hydroxyvitamin D3 level of 36.89 ng/mL (normal range, 8.0–51.9 ng/mL) (Fig. 1). In the spot urine test, the calcium/urinary creatinine ratio (mg%: mg%) was 1.31.
In a renal ultrasonography conducted because of persistent hypercalciuria, multiple small calyceal stones in both kidneys and a 0.6-cm stone in the left renal pelvis were newly observed, and hydronephrosis was absent (Fig. 3). Except for the dose of thiazide, the doses of the other drugs (calcium carbonate, magnesium, and calcitriol) were decreased. We carefully monitored the neonate for convulsions, and the serum calcium, phosphorus, magnesium, and urinary calcium excretion rate were checked regularly. On day 7 after readmission, calcitriol dose was decreased from 1.5 to 0.125 μg/day and the calcium dose was decreased to 20 mg/kg/day. In a blood test conducted 2 days later, the serum calcium level was found to be low (6.6 mg/dL). Consequently, the calcium carbonate dose was increased and maintained at 40 mg/kg. Due to hypercalciuria (spot urine calcium/creatinine ratio, 1.03), the thiazide dose was increased from 0.5 to 1.5 mg/kg, and the patient was discharged after we decided to control the doses in an outpatient setting.

(A) Kidney ultrasonography shows multiple tiny calyceal stones(white arrows) in right kidney. (B) A 0.6-cm stone filling the left renal pelvis(white arrow). (A,B) There was no evidence of obstructive uropathy.
In an outpatient blood test conducted on day 10 after discharge (when the patient was 3 months old), the serum calcium level was 7.5 mg/dL, the phosphate level was 7.6 mg/dL, and the spot urine calcium/creatinine ratio was 0.9. No neurological symptom was present. Hypercalciuria improved slightly during the administration of thiazide. However, hypocalcemia and hyperphosphatemia continued. Because of the persistent hypercalciuria and kidney stones, calcitriol administration was stopped. The patient had a history of neonatal hypocalcemic convulsions; hence, calcium carbonate was maintained at 40 mg/kg. The patient is currently being followed up and we are considering regulating the doses, with blood monitoring blood, urine tests, and renal ultrasonography.
This case was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital (approval number: 2017-11-008). Informed consent was obtained from both parents for the publishing of this case report.
Discussion
In the present case, a de novo novel variant of CaSR gene was confirmed in a neonatal patient who presented with convulsions that were caused by hypocalcemia. The patient also presented with hypoparathyroidism, hypocalcemia, and hypomagnesemia.
Generally, the causes of hypocalcemia within the first 72 hours after birth include preterm birth, fetal growth retardation, maternal diabetes, neonatal asphyxia, hypoparathyroidism, phosphorus overconsumption and hypomagnesemia [11,12]. Because genetic mutations are rare causes of hypocalcemia, blood tests to measure PTH, phosphate, magnesium, and vitamin D are conducted in neonatal and young patients with hypocalcemia [13,14]. Hypomagnesemia reduces parathyroid secretions and vitamin D synthesis, and it results in unresponsiveness or hyporesponsiveness in the organs on which PTH acts [15]. This is why the hypomagnesemia was corrected preferentially. Furthermore, the activating actions of CaSR can increase the renal excretion of calcium and the excretion of serum magnesium, thereby, decreasing parathyroid actions [16,17]. Thus, if hypocalcemia persists during the correction of hypomagnesemia and hypocalcemia or presents with hypoparathyroidism and hypercalciuria, activating mutations of CaSR gene should be considered.
Most activating mutations of CaSR gene have been reported to be missense mutations in patients with ADHH [8]. Hu et al. [10] reported a de novo mutation of CaSR gene in monozygotic twins. A variant of CaSR gene found in our patient (c.2474A>T [p.Tyr825Phe]) is an unreported de novo novel variant. The variant corresponds to the transmembrane-spanning domains. CaSR, which is composed of 1,078 amino acids, consists of a large extracellular domain comprising seven transmembrane-spanning domains and an intracellular tail and is coded by six exons of the CaSR gene that are located on chromosome 3q13.3–21 [3]. Although the patient in our report had a novel variant of CaSR gene, she showed the same clinical features (hypocalcemia, hyperphosphatemia, and hypercalciuria) noted in previously reported cases of CaSR gene mutations. If conservation scores are reliable in disease prediction, the c.2474A>T (p.Tyr825Phe) variant could be disease causing, as both Phastcon and GERP scores are high (0.994 and 5.89, respectively). Also, the patient's variant was not found in the control population and occurred at p.T825P, a similar location as in other pathogenic variants (p.A824P and p.T828N) [8]. Thus, the patient's variant is likely to be a pathogenic variant.
Regarding treatment, activating mutations of CaSR gene may cause the body to recognize low calcium concentrations as normal and, thus, influence parathyroid secretions [18]. Subsequently, the administration of calcium can aggravate hypercalciuria. The administration of calcium should be minimized in patients with an activating variant of CaSR gene; although calcitriol can be used, it can also cause hypercalciuria [19]. In particular, activating mutations of CaSR gene in patients with ADHH are sensitive to calcitriol, resulting in hypercalciuria after low doses of calcitriol. Therefore, hydrochlorothiazide (0.5–2.0 mg/kg/day) should be administered alongside, leading to the increased reabsorption of calcium in the renal tubules and, thus, a dose-sparing effect of calcitriol.
In the present case, renal stones were observed only 2 months after the administration of calcium and calcitriol. We planned to stop calcium carbonate and minimize the use of calcitriol to prevent severe hypercalciuria. The patient had a history of hypocalcemic convulsions and persistent hyperphosphatemia. It a reason that calcium carbonate could not be stopped. Only calcitriol was stopped, but the thiazide treatment was continued. Blood and urine tests along with renal ultrasonography will be regularly performed during the patient’s follow-up for the dosage monitoring and modification.
In conclusion, we report a de novo novel variant of CaSR gene (c.2474A>T [p.Tyr825Phe]) in a patient who presented with hypoparathyroidism with hypomagnesemia and hypercalciuria. A functional study regarding this variant is necessary.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.