Introduction
Puberty is the transition period between childhood and adulthood which takes place in several sequential stages controlled by neuroendocrine factors that regulate the onset and progression to sexual maturity. This neuroendocrine activity stimulates the secretion of the pituitary gonadotropins, i.e., follicle-stimulating hormones (FSH) and luteinizing hormones (LH), which are initially secreted pulsatile during the night and increase in amplitude and frequency throughout the day until reaching typical adult patterns [1]. This mechanism is known as activation of the hypothalamus-pituitary-gonadal axis, which stimulates the gonads to produce sex steroids (i.e., testosterone and estradiol, in males and females, respectively) inducing the appearance of secondary sexual characteristics and growth spurt. In the female, the breast bud appears and evolves until it becomes the classic breast of the adult woman passing morphologically from the 1st to the 5th stage of the Tanner classification. In the male, we observe an enlargement of the testicular volume over 4 mL, measurable using the Prader orchidometer, which consists of a series of ellipsoids which allow us to define the size of the male gonads by direct comparison. In 95% of healthy subjects of Caucasian origin, the breast bud appears in females between 8 and 13 years of age, while the increase of testicular volume in males occurs between 9 and 14 years of age [2,3]. In girls, the first menstruation is observed after on average around two and a half years after the appearance of the breast bud, while the pubertal spurt occurs between the 2nd and the 3rd stages of breast development. The puberty shoot occurs later in males, usually towards the 3rd or 4th stage, when testicle size reaches 12–15 mL. During the pubertal period, some males may experience swelling of the breast tissue, called gynecomastia, which is devoid of pathological significance. In both sexes, hair growth in the pubic area, known as "pubic hair," is not indicative of puberty because it is dependent on hormones secreted by the adrenal gland.
Clinical approach to delayed onset of puberty
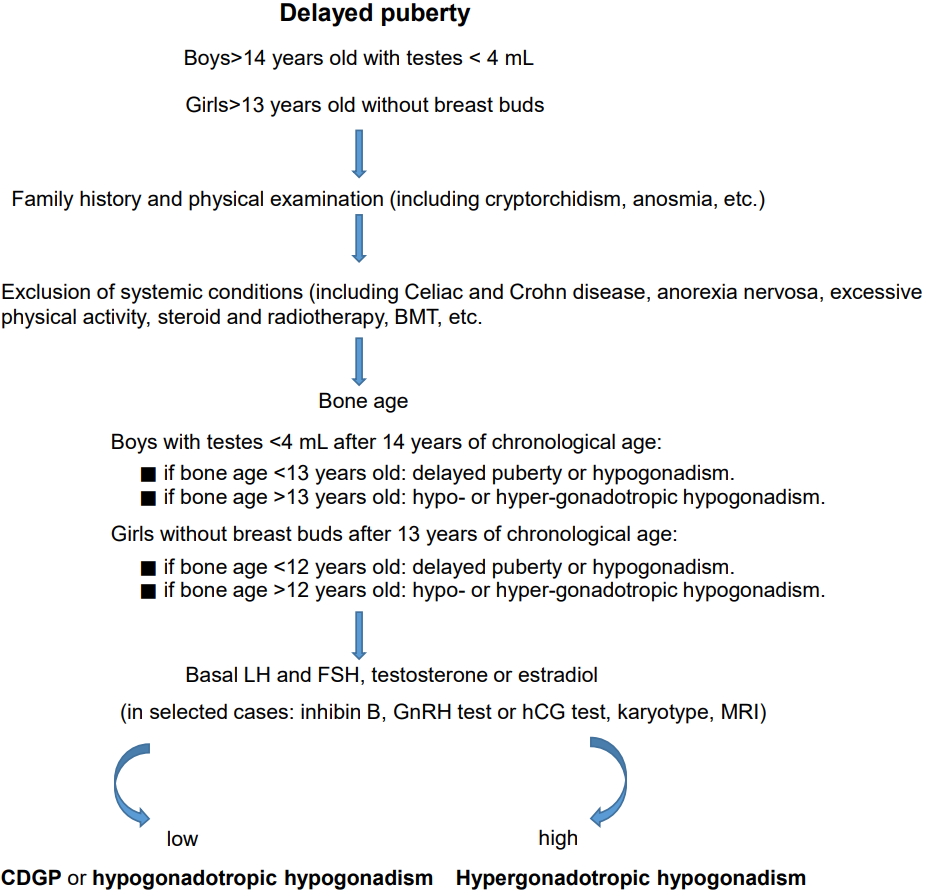
The clinician should follow the simple guidelines summarized in Fig. 1 when dealing with a subject with delayed onset of puberty.
Delayed puberty (DP) is considered as the absence of signs of sexual maturation by an age of more than 2–2.5 standard deviation values above the mean of the population. In reference to the Caucasian race, DP in the female is defined as the absence of breast bud at 13 years of age or a time lapse of more than 5 years from the occurrence of breast buds to the first menstruation, or no menstruation by age 16. In the male, DP is defined as lack of increase in testicular volume at 14 years of age or a time lapse of more than 5 years from the start to completion of genital growth. In a significant proportion of cases, DP represents the extreme end of the distribution of normal timing of puberty, rather than an overt pathology. The most frequent form, especially in males, is constitutional growth and puberty delay (CDGP), which is a non-pathological condition characterized by short stature and delayed skeletal maturation. After a thorough medical history, a correct auxological evaluation must be carried out with the determination of height, weight and growth rate and a clinical examination aimed at identifying particular signs, in addition to the evaluation of pubertal development and its progression. In about two-thirds of cases, CDGP is inherited from one of the parents [4]. A family history of DP is strongly indicative of CDGP (observed in 50%–75%) [4]. It is generally a transient condition with good prognosis both in terms of stature and future reproductive capacity. However, no consensus is reported on the question whether boys with DP are able to reach a final height appropriate for the mean target height [5,6]. Adolescents with CDGP may experience a peripubertal deceleration (i.e., slowing of growth rate) with delayed pubertal development and bone maturation compared to peers. The diagnosis is formulated by excluding other causes of pubertal delay such as organic, genetic, nutritional, or psychological causes. The presence of intestinal malabsorption should be excluded, especially celiac disease and Crohn disease, as well as subclinical hypothyroidism or cystic fibrosis. Excessive gymnastic-sporting activity may be responsible for pubertal delay, as observed in athletes undergoing prolonged and intense training.
Investigations are needed in cases showing more severe delay or clinical concerns. It is often clinically challenging to differentiate adolescents with CDGP from those with a form of HH, named isolated HH, which is usually a permanent condition [7]. Distinguishing between these conditions is especially difficult during initial evaluations because adolescents with both etiologies are often prepubertal on examination and have low levels of gonadotropins (LH and FSH). LH and FSH levels are low in CDGP because the hypothalamic-pituitary-gonadal axis has not yet matured enough to secrete pubertal levels of gonadotropin-releasing hormone (GnRH). In fact, a small percentage of boys with DP have a lifelong deficiency of puberty hormones such as LH and FSH, suggesting an isolated gonadotropin deficiency. However, neither baseline nor GnRH pituitary stimulated gonadotropin levels can serve to easily differentiate CDGP from permanent hypogonadotropic hypogonadism (HH). The majority of males with DP present short stature in comparison with their peers and retarded bone age. Some authors "prime" these patients by using small doses of sex steroids in order to carefully record a real growth hormone (GH) deficiency before starting GH therapy, in the event of low GH response to pharmacological stimuli.
If insufficient testosterone production in males or insufficient estrogen production in females is observed, associated with a blunted secretion of gonadotropins such as Kallmann syndrome (associated with reduction or absence of smell in 30%–50% of subjects) [8], or associated with GH deficiency, such as panhypopituitarism, a condition of HH may be suspected. An alteration of the transcription factors that regulate the development of the pituitary during embryogenesis, such as POUF-1, LHX-3, LHX-4, and HESX-1, including the appearance of multiple pituitary deficits may be involved [9,10], In these cases, it is necessary to perform a nuclear magnetic resonance spectroscopy, aimed at studying the hypothalamic-pituitary region, to exclude neoplasms or morphological alterations. Congenital HH may be diagnosed in males as cryptorchidism with or without micropenis during the mini-puberty of infancy (known as a transient activation of the hypothalamus-pituitary-gonadal axis within the first few months of life) [11] either in adolescence or in early adulthood when patients fail to show spontaneous pubertal development. Extreme thinness may well suggest the possibility of anorexia nervosa, while significant obesity may indicate genetic obesity, such as Prader Willi syndrome or Laurence-Moon-Bield syndrome. An absence of gonadotropin secretion is found in subjects receiving chemotherapy and radiation therapy for previous onco-hematological diseases. Hypothalamic and pituitary tumors such as adenoma, microadenoma and craniopharingioma may be associated with HH.
On the other hand, gonadal hormones may be associated with high levels of hypophyseal gonadotropins showing a form of hypergonadotropic hypogonadism. Females with primary ovarian failure have high gonadotropin levels because of lack of feedback of gonadal steroids on the pituitary gland. Turner syndrome is the most common cause of primary ovarian failure. Other conditions of primary ovarian failure include prepubertal surgical removal or irradiation of the ovaries for treatment of cancer, and autoimmune ovarian failure associated with other autoimmune diseases. Therefore, only in selected cases with DP is the use of dynamic tests recommended, such as the GnRH test to verify the response of the pituitary gonadotropins, the pelvic ultrasound to evaluate the uterus-ovarian size and the hand radiograph to verify skeletal maturation. Males with primary testicular failure have elevated gonadotropin levels due to the lack of testosterone feedback on the pituitary gland. This condition includes congenital bilateral anorchia ("vanishing testes") where no testes are found on surgical exploration and there is no testosterone response to human chorionic gonadotropin (hCG) stimulation. Moreover, chemotherapy, irradiation, surgical excision and infection in the prepubertal boy may result in testicular failure. In Klinefelter syndrome, which is the most common cause of testicular failure, puberty begins at the usual age and progresses normally, but testes are small and firm. Gynecomastia often occurs too.
In cases of high gonadotropin, it is necessary to perform a genetic analysis to highlight chromosomal alterations, such as Turner's syndrome in females and Klinefelter syndrome in males, which will result in a lack of pubertal progression and infertility problems in adulthood.
Differential diagnosis
The classification of pubertal delay requires an accurate medical history and a thorough objective examination including exclusion of dysmorphic syndromes and systemic diseases. Sometimes a physical examination alone is enough. Key findings are when the penis and testicles have not enlarged by age 14 in males and when the breast buds have not appeared by age 13 in females. Most boys with constitutional DP are shorter than their peers because of the delayed growth spurt. This condition is difficult to distinguish from isolated HH, a pathologic condition that requires treatment. Gonadotropic response to pharmacological stimuli, the evaluation of testicular volume as measured with the Prader orchidometer and the size of the uterus and ovaries as determined by pelvic ultrasound, may help in reaching a diagnosis. In girls, pelvic ultrasound examination is helpful when it reveals multicystic ovaries, a classic sign of early puberty. Recent studies indicate that inhibin B demonstrates the presence of testicular tissue in males and reflects follicular function in females. Inhibin B may provide a simple first-line test capable of identifying a subset of patients with DP who are highly likely to have CHH. Initial investigations should include an x-ray of the left hand to estimate bone age, which is usually delayed in relation to chronological age, suggesting that growth is as yet incomplete. Further investigations including genetic analysis and nuclear magnetic resonance spectroscopy may contribute to a more accurate definition of specific cases. Measurements of serum FSH and LH levels after GnRH will help to identify patients with hypergonadotrophic hypogonadism. Caution must be exercised in the interpretation of low gonadotropin concentrations, especially in patients under 12 years of age, because GnRH testing rarely clarifies whether an adolescent will progress in puberty or has a permanent defect [12].
Overnight spontaneous LH pulses are sensitive indicators for the onset of puberty [1]. Frequent samplings during the night have been proposed to determine the onset of puberty. Although this method could be useful in distinguishing CDGP from HH, it is considered to be an impratical routine diagnostic test [13].
Given the limited diagnostic value of basal pituitary gonadotropins, the GnRH test has been proposed as a useful method to accurately distinguish subjects with DP from HH patients. Unfortunately, the variability of the response and the overlap of LH levels with GnRH in subjects in the early stages of puberty do not ensure diagnostic accuracy [14].
GnRH agonists (GnRHa) appear to offer better discriminatory value than the GnRH test, according to the reported studies in the literature, including Nafarelin [15], Triptorelin [16], Buserelin [17], and Leuprolide [12].
The hCG test is based on the capacity to increase androgen production in Leydig cells after stimulation of LH receptors [18]. Neither hCG nor a combination of GnRH or GnRHa and hCG stimulation tests improve diagnostic sensitivity [19].
Finally, inhibin B which is secreted by Sertoli cells in the testis (males) and by granulosa cells in the ovary (females), significantly increases during puberty. Inhibin B may be considered a simple first-line test for the diagnosis of DP in males, but requires further investigations before indicating the same diagnosis in females [20].
Treatment
In cases of constitutional DP, the best course of action is patience and reassurance. Pediatric endocrinologists may offer a brief course of testosterone if psychological problems are exacerbated by the delay. In fact, the recommendation to treat CDGP is limited to prepubertal subjects older than 14 years of age with serious psychological distress, in particular due to bullying. However, once puberty has started, the administration of sex steroid hormones must be discontinued and the progression of puberty be allowed to take its course. In girls with CDGP, treatment with a very small amount of estradiol for up to 12 months is uncommon and should induce breast development early on. These treatments should be used cautiously to avoid any acceleration in skeletal maturation and the consequent risk of reducing final height. On the other hand, in the forms of hypo- or hyper-gonadotropic hypogonadism long-term hormone replacement treatment is recommended. Girls usually start on a low dose of estrogen administered orally with tablets or through transdermal patches or gel. Within one year, progesterone is added to induce the menstrual cycle and increase bone density during puberty. Boys usually start on a low dose of testosterone administered orally with tablets or through injections to initiate secondary sexual development. Once growth and puberty are almost completed, then testosterone injections, implants, patches or gel are used for ongoing lifelong hormone replacement. Long-acting, slow-release testosterone is currently available in the form of intramuscular injections administered once every three months.
Effective treatment such as pulsatile GnRH or gonadotropin therapy (hCG alone or combined with FSH) is available not only for inducing virilization or estrogenization, but also for successful development of fertility.
On the other hand, treatment with appropriate sex steroid replacements for adolescents with a permanent cause of DP such as hypo- and hypergonadotropic hypogonadism should be initiated at the usual age of puberty.
Conclusions
CDGP subjects have a late but otherwise normal puberty which occurs spontaneously, while hypogonadic patients do not initiate spontaneous pubertal development. In many patients, routine initial clinical evaluation cannot distinguish CDGP from IHH with certainty. Moreover, 10% of hypogonadic hypogonadism subjects reverse later in life because of the clinical overlap between CDGP and hypogonadic patients [21].
The spontaneous onset of steadily progressive puberty by 18 years of age must be considered the gold standard for differentiating CDGP from HH. Clinicians often cannot distinguish CDGP from isolated HH, with a definitive diagnosis suspended until lack of spontaneous puberty by age 18 is confirmed. However, the ability to make a correct diagnosis early on carries clinical implications. The lack of pubertal progression for more than two years after spontaneous onset at the appropriate age is indicated by a failure to achieve menarche from the onset of thelarche in girls, and in boys the attainment of adult size of testes from 4 mL for more than 5 years. Pediatric Endocrinologists should routinely propose GnRH or GnRH agonist tests as early discriminators to obtain a correct diagnosis quickly and thus avoid anxiety in patients and their families. Further large-scale studies are required to confirm that basal inhibin B may offer a simple, discriminatory test.
The clinician should follow the simple guidelines summarized in Fig. 1 when dealing with a subject with delayed onset of puberty.