Congenital hyperinsulinism: diagnostic and management challenges in a developing country – case report
Article information

Abstract
Management of congenital hyperinsulinemia of infancy (CHI) is challenging. A 4-month-old female infant with persistent hypoglycemia and elevated insulin levels was diagnosed with CHI. Gallium-68 DOTANOC positron emission tomography/computed tomography (PET/CT) scan (68Ga-labeled [1,4,7,10-tetraazacyclododecane-N,N’,N’’,N’’’-tetraacetic acid]-1-NaI3-octreotide) demonstrated focal disease in the body of the pancreas. Genetic studies indicated paternal inheritance, making focal disease likely. She was started on diazoxide therapy with partial improvement in blood glucose levels. Due to a suboptimal response to diazoxide and the likelihood of focal disease amenable to surgery, a laparoscopic subtotal pancreatectomy with preservation of the head of the pancreas was performed. The biopsy demonstrated diffuse hyperplastic pancreatic islet cells on immunohistochemistry, indicative of diffuse rather than focal disease. Paternal inheritance is a recognized indicator of focal disease. Gallium-68 DOTANOC PET/CT scan is the only available imaging modality in South India as 18F-L-dihydroxyphenylalanine (DOPA) PET/CT scan is not available at present. A laparoscopic approach reduces the postoperative recovery time and morbidity in such patients. The absence of 18F-L-DOPA PET/CT scan and the limited supply of diazoxide makes the management of this complex condition more challenging in developing countries.
Introduction
Congenital hyperinsulinemia of infancy (CHI) is a major cause of hypoglycemia in the neonatal period and early infancy. It has also been referred to as persistent hyperinsulinemic hypoglycemia of infancy, primary islet cell hypertrophy and nesidioblastosis. It is characterized by inappropriate secretion of insulin in relation to blood glucose levels and can result in recurrent and persistent hypoglycemia in neonates and infants [1].
Early recognition of CHI and maintenance of euglycemia is necessary to minimize neurologic damage which can result from recurrent and/or prolonged episodes of hypoglycemia [2]. CHI has a genetic basis and there are 2 histological subtypes: diffuse and focal [3]. The identification of focal disease by 18F-L-dihydroxyphenylalanine (DOPA) positron emission tomography/computed tomography (PET/CT) scan may help prompt removal of the focal lesions, thereby possibly rendering cure. However, 18F-L-DOPA PET/CT scan is not available in South India, and hence, some centres use Gallium-68 DOTANOC PET/CT scan (68Ga-labeled [1,4,7,10-tetraazacyclododecane-N,N',N'',N'''-tetraacetic acid]-1-NaI3-octreotide) although it is not as accurate as 18F-L-DOPA PET/CT scan [4].
Medical options for treatment of diffuse CHI include diazoxide and octreotide therapies. Diazoxide is the first line of therapy and is effective in maintaining normoglycemia in those patients who are responsive to diazoxide but unfortunately. Diazoxide is not freely available in India, making medical management very challenging. Surgery is considered as a treatment option if focal disease has been identified on imaging or in medically unresponsive diffuse disease [1].
Case report
A 4-month-old female infant was seen with clinical features of persistent hypoglycemia. She was born at 40-week gestation by lower segment cesarean section and weighed 3.9 kg. Her mother did not have diabetes mellitus and she was not on any medications. There was hypoglycemia necessitating dextrose infusions from the second to eleventh day of life. We do not have calculations of the glucose delivery rate in mg/kg/min administered to the baby in that nursing home at that time. The baby was discharged after blood glucose ‘normalized.’ There were 3 episodes of generalized tonic-clonic seizures at 33 days of age necessitating admission to the local medical college hospital, where the baby received intravenous dextrose, hydrocortisone and an unsuccessful trial of Nifedipine. The baby did not have any features of macrosomia. The parents had been adding one sugar cube to each top-up feed of formula following a breast feed but the blood glucose still remained low.
From a neurodevelopmental point of view, her ability to support her head had not fully developed at 4 months. However, she did have social smile and was fixing and following. There was no family history of either diabetes mellitus or CHI.
Investigations at the age of 4 months when she was assessed in the pediatric endocrinology clinic were demonstrated in Table 1.
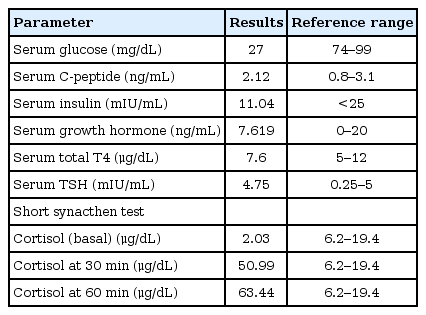
Investigations of 4-month-old female infant
Gallium-68 DOTANOC PET/CT scan (Discovery VCT PET scanner, GE Healthcare, Chicago, IL, USA) demonstrated diffuse increased uptake of 68Ga DOTANOC in the body of the pancreas (Fig. 1). A screening echocardiogram prior to commencing diazoxide therapy was negative for structural disease. She was also prescribed hydrochlorothiazide in line with current recommendations to prevent fluid overload in patients on diazoxide. Early weaning was recommended as she was 4 months old.
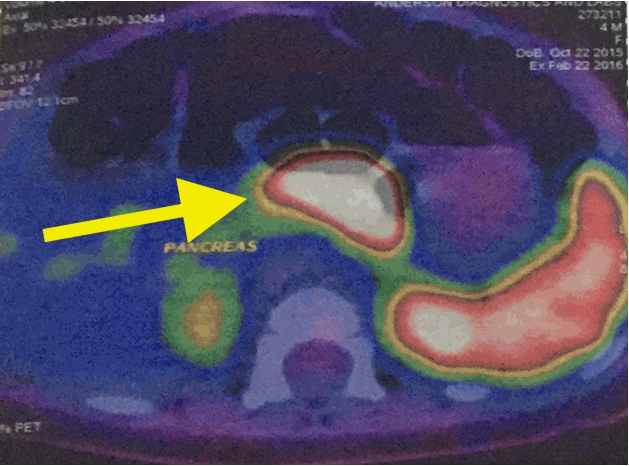
Gallium positron emission tomography scan demonstrated increased uptake of 68Ga DOTANOC in the body of the pancreas (yellow arrow).
She was commenced on diazoxide therapy with transient improvement in blood glucose levels on an initial total daily dose of 5 mg/kg/day. After 2 days, the dosage of diazoxide was increased to 15 mg/kg/day with some improvement in the blood glucose levels (60–100 mg/dL). Her parents reported that she became more alert and interactive. Genetic tests performed on the infant and her parent's samples at Genetics Laboratory, Royal Devon & Exeter Hospital, United Kingdom confirmed that she had inherited heterozygous ABCC8 mutation from her father which is usually suggestive of focal disease. Sequence analysis identified a previously reported heterozygous paternally inherited missense ABCC8 mutation (p.R1421C [c.4261C>T]) [5]. There have been no published reports of CHI in India caused by this mutation.
In view of the suboptimal response to diazoxide therapy, she was referred to the paediatric surgeons who performed a laparoscopic subtotal pancreatectomy with preservation of the head of the pancreas. The whole excised specimen (Fig. 2) was sent for histopathology. The histopathology was suggestive of diffuse hyperplastic pancreatic islet cells suggestive of diffuse CHI (Fig. 3).
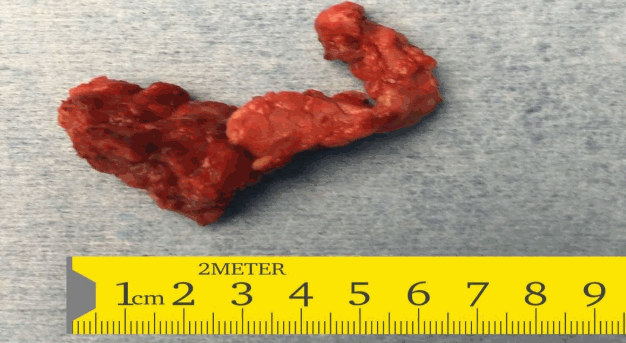
Resected specimen of subtotal pancreatectomy demonstrating body and tail of pancreas.

Histopathology section in high power view focusing the pale acini (H&E, ×200), demonstrates diffuse uptake of hematoxylin throughout acini by the beta cells suggestive of diffuse disease characterised by hypertrophied beta cells with abnormal large nucleus.
Postoperatively, the infant was started on enteral feeds within 12 hours of surgery. There was initial hyperglycemia which gradually resolved over 48 hours. Apart from 2 episodes of hypoglycemia in the postoperative period, she made an uneventful recovery and was discharged on the 5th postoperative day. The family was counselled to look for features of hypoglycemia, monitor blood glucose at home and to continue breast feeding with a top up feed with formula milk if the blood glucose dropped below 60 mg/dL.
One week following the surgery, she was subjected to a controlled fast. Three hours after the fast commenced, the blood glucose dipped to 58 mg/dL. This suggested that there is still some residual hyperinsulinism. Currently, the baby is breast fed and has been fully weaned. Blood glucose levels are between 70–110 mg/dL on frequent feeds and the baby has not been recommenced on diazoxide. Further pancreatic surgery might be warranted if the hypoglycemia recurs, but a balance has to be struck between preventing hypoglycemia and rendering the infant diabetic with a more radical resection of the pancreas.
Institutional Review Board approval for this case was not required as this case report described the treatment of a single patient. Parent's written consent has been obtained for this publication.
Discussion
CHI commonly presents with symptomatic hypoglycemia in the neonatal period. Studies have shown that 75% of patients develop symptoms within the first three months of life [6]. Rarely, a child may be picked up with CHI above the age of one year [1]. Most of the babies diagnosed with CHI are macrosomic when they are born [1].
Genetically, this is felt to be a heterogeneous condition [7,8]. There is a wide spectrum in clinical presentation with mild symptomatic hypoglycemia at the milder end to life-threatening hypoglycemia at the severe end of the spectrum. The response of babies with CHI to medical treatment and surgery may also be variable [7].
The main goal of therapy in CHI is to prevent symptomatic hypoglycemia which may manifest as seizures, lethargy, or even coma in the most severe cases. This will then prevent long-term sequelae like epilepsy, deficits in cognition and in some cases, microcephaly [2].
Pharmacologic therapy for CHI involves treatment with diazoxide (a specific ATP-dependent potassium [KATP] channel agonist), somatostatin analogues (octreotide), or calcium channel blockers (nifedipine) [1].
Diazoxide is the first-line therapy. It works by blocking the sulfonylurea receptors on the beta cells of the pancreas, resulting in opening of the KATP channels, and decreasing insulin release. Diazoxide is administered at doses of 5 to 15 mg/kg per day in three divided doses. However, it is not readily available in India.
The imaging modality of choice to diagnose focal CHI is 18F-L-DOPA PET/CT scan [1,9]. 18F-L-DOPA is the acronym for 6-[18F]-L-fluoro-L-3, 4-dihydroxyphenylalanine. However, this imaging modality is currently not available in South India and is available only in a limited number of institutions around the world. The PET/CT scan usually will demonstrate a uniform uptake of 18F-L-DOPA throughout the pancreas in cases of diffuse CHI whereas in focal CHI, the uptake may be noted only in particular foci of disease within the pancreas. The detection limit of the scan is 105 to 106 cells or a diameter of 1 mm [9]. Its use will reduce but not eliminate the number of near-total pancreatectomies in infants with focal CHI. The 18F-L-DOPA PET/CT scan does not always accurately demonstrate the site or size of the lesion.
Gallium-68 DOTANOC PET/CT scan is not a reliable imaging modality for distinguishing between focal and diffuse CHI. There is limited evidence in medical literature supporting its use in CHI [4].
Pancreatectomy is usually considered as the last option in management of CHI when medical measures have been unsuccessful in maintaining prolonged euglycaemia. Paternal inheritance of the mutation has a strong association with focal disease [1,3] and hence, we opted to treat the patient with subtotal pancreatectomy. Not all patients with a paternally inherited KATP channel mutation have focal uptake by 18F-L-DOPA PET/CT scan, and some of these actually show diffuse histology [10,11]. Banerjee et al. [10] reported that 31% of patients with a paternal monoallelic mutation showed diffuse uptake on 18F-L-DOPA PET/CT scan. A sensitivity of 94% and a specificity of 100% have been reported when comparing 18F-L-DOPA PET/CT scan and histology according to an international questionnaire and data published from the USA [12,13]. Focal lesions, even those that have been localized before surgery, can sometimes be difficult to fully resect during surgery. A frozen section of the pancreas during the surgery performed by a dedicated histopathologist is necessary for adequate resection of the focal lesion. Till recently, the most commonly performed surgical procedure was a subtotal pancreatectomy which would have removed 95% of the pancreas. This was successful in many cases but some infants went on to require a second surgery to resect the remnants of the pancreas [1]. Insulin-dependent diabetes mellitus is a relatively common complication of near total pancreatectomy and is known to sometimes occur many years after pancreatectomy [14]. Near-total pancreatectomy may be the best available management option for diffuse CHI [15] while surgical resection may be curative in focal CHI.
Surgeons used to perform open pancreatectomy in babies with CHI but recently, laparoscopic pancreatectomy has been successful [16]. Data suggest that laparoscopic pancreatectomy for medically unresponsive CHI is feasible and safe. Laparoscopic pancreatectomy is associated with less intraoperative complications like blood loss and associated morbidity, and can even reduce the duration of hospital stay [17].
Samples for genetic analysis from developing countries are analyzed gratis in the United Kingdom but the limited availability of diazoxide and the absence of 18F-L-DOPA PET/CT scan make the management of these complex patients more challenging.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.