A 1-month-old infant with chylomicronemia due to GPIHBP1 gene mutation treated by plasmapheresis
Article information

Abstract
Chylomicronemia is a severe type of hypertriglyceridemia characterized by chylomicron accumulation that arises from a genetic defect in intravascular lipolysis. It requires urgent and proper management, because serious cases can be accompanied by pancreatic necrosis or persistent multiple organ failure. We present the case of a 1-month-old infant with chylomicronemia treated by plasmapheresis. His chylomicronemia was discovered incidentally when lactescent plasma was noticed during routine blood sampling during a hospital admission for fever and irritability. Laboratory investigation revealed marked triglyceridemia (>5,000 mg/dL) with high chylomicron levels. We therefore decided to perform a therapeutic plasmapheresis to prevent acute pancreatitis. Sequence analysis revealed a homozygous novel mutation in exon 4 of GPIHBP1: c.476delG (p.Gly159Alafs). Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) stabilizes the binding of chylomicrons near lipoprotein lipase and supports lipolysis. Mutations of GPIHBP1, the most recently discovered gene, can lead to severe hyperlipidemia and are known to make up only 2% of the monogenic mutations associated with chylomicronemia. The patient maintains mild hypertriglyceridemia without rebound after single plasmapheresis and maintenance fibrate medication so far. Here, we report an infant with chylomicronemia due to GPIHBP1 mutation, successfully treated by plasmapheresis.
Introduction
Severe hypertriglyceridemia is diagnosed when the plasma triglyceride (TG) concentration exceeds 885 mg/dL1). Chylomicronemia, formerly known as type 1 hyperlipoproteinemia, is a type of severe hypertriglyceridemia characterized by the accumulation of chylomicrons in fasting plasma2).
Chylomicronemia usually presents in infancy or childhood with episodes of eruptive xanthomas on the trunk and extremities, lipaemia retinalis, recurrent abdominal pain, acute and/or chronic pancreatitis and hepatosplenomegaly3). It is often discovered incidentally by lactescent plasma during routine blood sampling. The most serious manifestation is pancreatitis, which is associated with 5%–6% overall mortality, and the highest mortality rate is related to the development of pancreatic necrosis or persistent multiple organ failure4).
Familial chylomicronemia has a monogenic basis with autosomal recessive pattern and arises from inactivating mutations in genes that encode key check-point molecules in lipolysis such as lipoprotein lipase (LPL), apolipoprotein C2 (APOC2), apolipoprotein A5 (APOA5), lipase maturation factor 1 (LMF1), or glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1)56).
GPIHBP1 binds LPL and translocates the enzyme to the luminal surface of capillary endothelial cells, thus supporting lipolysis7). GPIHBP1 mutations may disturb lipolytic processing, which leads to severe hypertriglyceridemia, and to date this disturbance has been reported in only 26 families891011).
The mainstay of treatment is a restriction of fat intake to less than or equal to 15% of total energy intake, and especially infants are given medium chain triglycerides (MCTs) milk, which are not incorporated into chylomicrons12). However, in cases with very high TG levels, dietary treatment may be insufficient for preventing pancreatitis, and nonpharmacological treatment, such as plasma exchange, becomes necessary.
Here, we report a 1-month-old infant with chylomicronemia due to a GPIHBP1 mutation and how he was treated by extracorporeal methods.
Case report
A 40-day-old male infant, born at 39 weeks and 3.3 kg by caesarean section without perinatal complications, was hospitalized with fever and irritability for 1 day. His height was 56 cm (25th–50th percentile) and weight was 4.6 kg (5th–10th percentile) with absolute breastfeeding. When a blood sample was taken for evaluation, physicians noticed lactescent plasma. He was then referred to the pediatric endocrinology clinic. He was the only child, and his family history was unremarkable. Physical examination did not reveal specific findings for dyslipidemia such as xanthomas and hepatosplenomegaly. His vital signs were stable with mild fever and mild tachycardia (blood pressure, 85/45 mmHg; heart rate, 140 beats/min; respiratory rate, 35 breaths/min; body temperature, 37.8℃). Laboratory investigation revealed pronounced hypertriglyceridemia (>5,000 mg/dL; normal range, 50 to 150 mg/dL), and hypercholesterolemia with total cholesterol at 988 mg/dL (normal range, 120 to 200 mg/dL), high-density lipoprotein cholesterol at 390 mg/dL (normal range, 40 to 75 mg/dL), and low-density lipoprotein cholesterol at 9 mg/dL (normal range, 70 to 160 mg/dL). Amylase and lipase levels were normal. Serum albumin was slightly lower than normal (3.2 g/dL; normal range, 3.8 to 5.4 g/dL), but there was no proteinuria or clinical symptom. The analysis of lipoproteins showed high chylomicron at 24.0% (normal range, 0–0.4), low alpha lipoprotein at 0.4% (normal range, 15.6–39.1), and low beta lipoprotein at 18.5% (normal range, 37.2–64.0). Primary chylomicronemia was diagnosed on this basis.
The first therapeutic measure was to give a diet free of long chain fatty acids by means of MCTs milk. In addition, therapeutic plasmapheresis was necessary to manage the high risk of pancreatitis. Therapeutic plasma exchange (Spectra optia, Thermo BCT Inc., Lakewood, CO, USA) was carried out using fresh frozen plasma and 20% albumin. Body weight was 4.6 kg and the patient's estimated blood volume was 345 mL. The blood flow rate was 6 mL/min and total procedure time was 150 minutes (removed volume, 373 mL; replaced volume, 318 mL) (Table 1). Anticoagulation was also performed by heparin bolus (30 IU/kg). During the procedure, vital signs, bleeding tendency, and electrolytes were carefully monitored, and there were no significant side effects such as hypovolemic shock. The patient tolerated the procedure, and blood samples were collected before and 24, 48, and 72 hours after the procedure to assess the TG rebound. Immediately after plasmapheresis, the TG level had dropped to 2,125 mg/dL and then gradually decreased to 1,230 mg/dL, 465 mg/dL, and 444 mg/dL after 24 hours, 48 hours, and 72 hours, respectively (Fig. 1). In addition, we started fibrate medication (Bezalip 30 mg/kg per day) and MCT oil. The patient was discharged 6 days after admission, and he continues to be regularly followed-up in the outpatient clinic to monitor his lipid profiles. For now, he is 1 year old with normal growth parameters; his height is 77 cm (25th–50th percentile) and weight is 10.2 kg (25th–50th percentile). The TG level stayed below 700 mg/dL during 1 year with same fibrate dosage without complications.

Plasma exchange technical parameters in 1-month-old infant with chylomicronemia
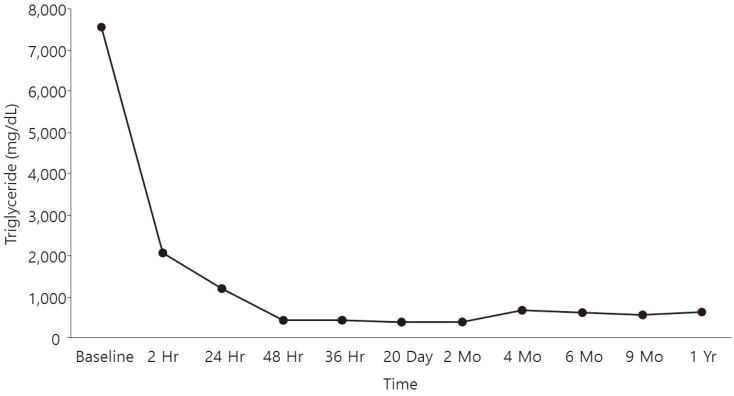
Plasma triglyceride and cholesterol levels before and after plasma exchange.
To identify the cause of chylomicronemia, genetic direct sequencing was performed. During the genetic investigation, a novel homozygous mutation was revealed in exon 4 of GPIHBP1 (c.476delG, p.Gly159Alafs), the gene that stabilizes the binding of chylomicrons near LPL and supports lipolysis (Fig. 2). Furthermore, genetic sequencing was performed for both parents, and his father harbored a heterozygous mutation, whereas, his mother had no mutation in GPIHBP1.
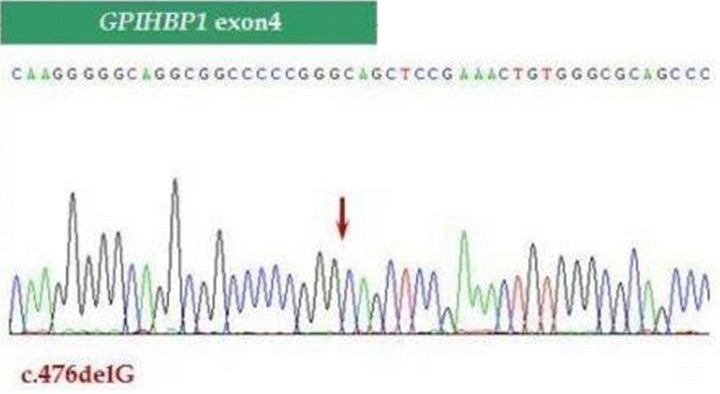
The mutation of nucleotide sequence in GPIHBP1 gene in patient.
Discussion
In this study, we have described a 1-month-old infant who was incidentally found to have chylomicronemia resulting from a GPIHBP1 mutation and treated by plasma exchange. Patients with TG levels greater than 885 mg/dL should be investigated by a step-wise approach. Secondary causes such as uncontrolled diabetes, hypothyroidism, poor diet, alcohol use, nephrotic syndrome, obesity, and a variety of medications can be responsible for severe hypertriglyceridemia in children13). If there is no secondary cause, monogenic factors should be considered. LPL deficiency is the most common cause of monogenic chylomicronemia. In the past, biochemical studies have been used to determine LPL or APOC2 activities, but nowadays genetic sequencing is performed for diagnosis. Patients with monogenic chylomicronemia show homozygosity or compound heterozygosity for loss-of-function mutations in causative genes such as LPL, APOC2, APOA5, LMF1, and GPIHBP114). Mutations in GPIHBP1, the most recently discovered gene, make up only 2% of monogenic mutations2). In the case at hand, the patient had a homozygous GPIHBP1 mutation and his father had a heterozygous mutation. Therefore, we can suppose that one allele was inherited from his father, and the other was from de novo mutation. Also, there was a possibility of paternal uniparental disomy as well. To our knowledge, only a few cases of infant monogenic chylomicronemia due to GPIHBP1 mutations have been reported. One was a 2-month-old infant homozygous for GPIHBP1 deletions who was treated with a low-fat formula diet8). The other was a 6-month-old infant with novel biallelic mutations in GPIHBP1 who improved on MCT-rich formula15). The case at hand is therefore the youngest case with GPIHBP1 mutations who has been treated by plasmapheresis reported so far.
The mainstay of chylomicronemia therapy is an extremely low-fat diet, and especially in infants supplemental MCTs provide additional calories without raising circulating TG levels. However, the long-term effects of diets with high levels of MCTs remain unknown15). In addition, traditional lipid-lowering medications, such as fibrates, niacin and statins, are minimally effective in patients with primary chylomicronemia, especially patients with complete loss-of-function mutations in the lipolytic pathway2). Although there are few studies about efficacy and safety of fibrates in children with hypercholesterolemia, Becker et al.16) reported fibrate is an alternative, safe and effective therapeutic choice for treatment of severe familial hypercholesterolemia in children. Therefore, we prescribed fibrate medication (Bezalip, 30 mg/kg per day) to reduce TG level definitely after single plasmapheresis.
Plasmapheresis or plasma exchange with direct removal of TG has been used, and plasma exchange is mentioned in the category III of the American Society for Apheresis 2010 guidelines (recommendation grade 2C, type of evidence II-I) for the treatment of hypertriglyceridemia17). In infant patients, Stefanutti et al.18) have reported successful plasmapheresis in a 25-day-old, 3.9-kg male with chylomicronemia whose TG level reached 38,000 mg/dL. Also Stefanutti et al.19) reported a 3-month-old infant with chylomicronemia treated with plasma exchange to avoid acute pancreatitis, whose TG level dropped dramatically within 72 hours (Initial TG level was 167.00 mmol/L, 11.68 mmol/L after 72 hours after plasma exchange). In comparison with conservative management of chylomicronemia in a 2-month infant reported by Rios et al.8) (Initial TG level was >25,000 mg/dL, 397 mg/dL after 12 days), the use of plasma exchange showed effective removal of TG level. However, small clinical trials comparing plasmapheresis with conservative management have shown that plasmapheresis has no overall benefit4). In addition, the TG concentration may return to the preapheresis values after plasmapheresis and there are no precise guidelines for therapeutic plasmapheresis in chylomicronemia. In our case, the TG level was severely raised, to above 5,000 mg/dL, and we opted for a drastic management strategy, therapeutic plasmapheresis, to prevent the acute pancreatitis. Fortunately, this reduced the TG level dramatically without complications, to below 400 mg/dL, and it stayed below 700 mg/dL for 1 year after only single plasmapheresis. Therefore, plasmapheresis may be an effective management strategy in infant patients with chylomicronemia, even though this is an invasive and risky procedure.
Recently, a new therapeutic approach has emerged with lomitapide (microsomal triglyceride transfer protein inhibitor). This drug is currently approved in North America and Europe for the treatment of homozygous familial hypercholesterolemia and has been shown to reduce triglyceride levels by 30%–40%20). However, lomitapide is only recommended for patients with pancreatitis associated with severe hypertriglyceridemia and without active hepatitis20). In addition, the safety of this drug for infant patients has not been proven. Further clinical trials in younger children are needed.
In conclusion, we report the youngest case of chylomicronemia due to GPIHBP1 mutation that has been successfully treated by plasmapheresis. Accordingly, this approach may be effective in infant patients with severe chylomicronemia.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.