Systemic primary carnitine deficiency with hypoglycemic encephalopathy
Article information

Abstract
Acute hypoglycemia in children is not an uncommon disease that can be encountered in the Emergency Department. Most cases of childhood hypoglycemia are caused by ketotic hypoglycemia due to missed meals. Often, hypoketotic hypoglycemia can also occur, which suggests hyperinsulinemia or a defect in fatty acid oxidation. Carnitine is essential for long chain fatty acids transfer into mitochondria for oxidation. We present a case of systemic primary carnitine deficiency who presented with seizures due to hypoketotic hypoglycemia.
Introduction
Carnitine takes an important role in the transfer of long-chain fatty acids across the inner mitochondrial membrane. Long-chain fatty acids are required for energy production during the fasting state. This transfer needs enzymes and transporters that deposit carnitine within the cell. The organic cation/carnitine transporter 2 (OCTN2) plays a key role in carnitine oral absorption, tissue distribution, and renal reabsorption. Deficiency in OCTN2 carnitine transporter leads to systemic primary carnitine deficiency (CDSP). It is characterized by decreased carnitine accumulation in tissues and increased losses of carnitine in the urine. Patients with carnitine deficiency may present with hypoketotic hypoglycemia and other symptoms. We present a case of CDSP confirmed by identification of a mutation of the Solute Carrier Family 22 Member 5 (SLC22A5) gene that encodes the OCTN2 carnitine transporter.
Case report
A previously healthy 3-year-old girl was admitted to the Emergency Department (ER) because of seizures and drowsiness. Three days before the ER visit, she had respiratory symptoms and her oral intake had been poor. The day before the visit, she had a fever and had taken medicines including Cefcapene Pivoxil HCl Hydrate (FLOMOX Fine granules for children 100 g, Ildong, Seoul, Korea) 2 doses. Her mother said that she had been relatively good in daytime and went to bed supperless. About 5 o'clock in the morning, one hour before the ER visit, she woke up with cold sweating and became irritable and drowsy. Generalized tonic-clonic type seizures developed en route to the hospital. She was born by Caesarean at full-term age and a weight of 3.8 kg. She was the first-born to nonconsanguineous parents. Her growth and developmental status was appropriate for the same age. Her mother said that newborn screening test and past medical history was unremarkable. She has a younger sister with normal growth development and no other symptoms or signs.
At the ER, physical examination findings included her height 104 cm (75th–90th percentile), weight 16.5 kg (75th percentile), body mass index 15.26 kg/m2 (50th percentile), heart rate 110/min, respiratory rate 24/min, body temperature was 37.7℃. She was in a state of tonic-clonic seizures and conscious level was deeply drowsy. Head and neck examination showed slight dried tongue, normal tympanic membrane and no palpable neck lymph nodes. On examination of the chest, breath and heart sounds were normal. Abdominal examination showed normal bowel sounds, without tenderness on palpation, and not apparent hepatosplenomegaly. There was no edema on extremity or rashes on the skin. Laboratory finding showed the following results: white blood cell 30.5 ×103/µL (reference range [RR], 5.5–15.5 ×103/µL), hemoglobin 11.8 g/dL (RR, 11.5–13.5 g/dL), platelet count 459 109/L (RR, 150–450 109/L), erythrocyte sedimentation rate 25 mm/hr (RR, 0–20 mm/hr), C-reactive protein 4.23 mg/dL (RR, 0.0–0.5 mg/dL), aspartate aminotransferase 47 U/L (RR, 13–35 U/L), alanine aminotransferase 16U/L (RR, 7–35 U/L), total cholesterol 156 mg/dL (RR, 150–250 mg/dL), triglyceride 48 mg/dL (RR, 50–130 mg/dL), free fatty acid 740 µEq/L (RR, 300–480 µEq/L), sodium 135 mEq/L (RR, 136–142 mEq/L), potassium 3.4 mEq/L (RR, 3.8–5.0 mEq/L), phosphorus 6.6 mg/dL (RR, 4.0–7.0 mg/dL), calcium 9.8 mg/dL (RR, 9.0–11.0 mg/dL), magnesium 3.1 mg/dL (RR, 1.3–2.1 mg/dL), blood glucose 14 mg/dL (RR, 70–110 mg/dL), blood ketone negative. Arterial blood gas analysis showed that pH 7.42, pCO2 29.0 mmHg, HCO3- 18.5 mmol/L. The level of insulin was 0.2 µIU/mL (RR, 4–24 µIU/mL), cortisol >62.80 µg/dL (RR, 6.7–22.6 µg/dL), ACTH 1,777.0 pg/mL (RR, 7.2–63.3 pg/mL). Blood ammonia was 136 µmol/L (RR, 9–35 µmol/L). Urine organic acid analysis showed that 4-Hydroxypheylacetic acid 73.3 mmol/mol ceatinine (RR, 0.0–69.9 mmol/mol ceatinine). Plasma amino acid analysis revealed no prominent abnormality. Urinalysis showed that ketone 2 positive but no proteinuria or hematuria. Urine microscopy revealed 0–2 /mm3 white cells and 0–2 /mm3 red cells. Chest radiography showed no cardiomegaly and no other significant abnormalities. The cerebral spinal fluid (CSF) study showed that white blood cell 0/µL, red blood cell 0–2 /high power field, glucose 27 mg/dL, protein 17.9 mg/dL (RR, 12–60 mg/dL), Herpes simplex type I polymerase chain reaction negative, enterovirus culture negative. The CSF pressure could not be accurately measured. Brain magnetic resonance imaging done on second hospital day showed bilateral symmetrical areas of hyperintensity on diffusion weighted imaging with a low apparent diffusion coefficient value in the thalami, parietal, and temporal lobes, and it was consistent with cytotoxic edema, which can be seen in hypoglycemic encephalopathy (Fig. 1).
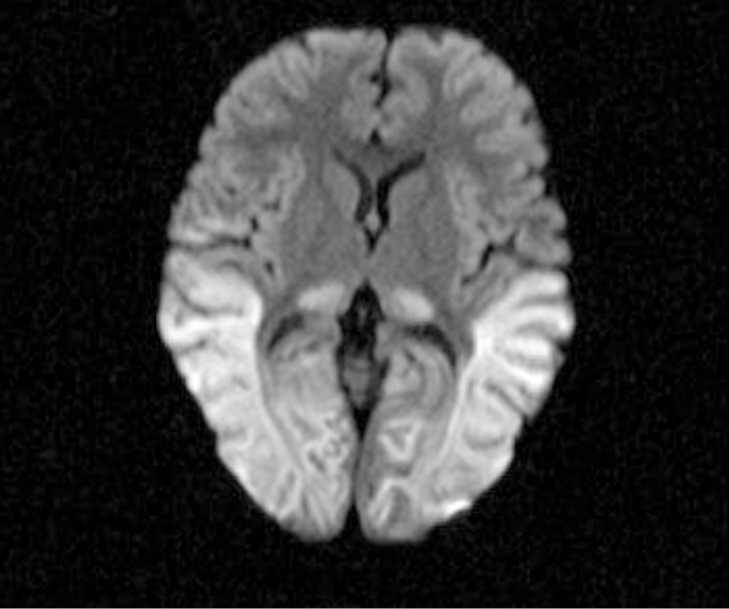
Diffusion weighted image shows swollen, markedly hyperintense white matter in the parietal, occipital lobes.
For all the anticonvulsants treatment such as midazolam, phenobarbital and phenytoin, her mentality was stupor and seizures were continued. Intubation and mechanical ventilation was conducted. Despite 10% glucose bolus and continuous infusion at a rate of 15 mg/kg/min with hydrocortisone at a dose of 5 mg/kg/day injection, blood glucose was stabilized above 70 mg/dL after 6 hours from initiation. Empirical L-carnitine solution 55 mg/kg/day divided 3 times was started on the second day. She could be extubated on the third day, but focal seizures developed and progressed. On the fifth day, she was reintubated and midazolam continuous infusion was done. She was seizures-free state on the ninth day, so the anticonvulsants could be tapered but she was kept on phenytoin, phenobarbital, topiramate and oxacarbazepine. Her visual fixation and tracking were disabled. The locomotion and transitional movements such as supine to sit were severely impaired. She was transferred to rehabilitation medicine on the 35th day.
We wonder the etiology of her hypoglycemia because the hospital course was extraordinarily severe and a negative serum ketone was also unusual. A few weeks later, we confirmed the initial serum carnitine level done on the third day was very low (<0.01 µmol/L). We consider primary or secondary carnitine deficiency by other metabolic disorders. The association of exposures to antibiotics containing pivalic acid and hypoglycemia was reported on CDSP. So we conducted gene analysis to confirm CDSP.
Analysis of SLC22A5, the gene that encodes the OCTN2 transporter, identified the c.396G>A(p.W132X) and c.539A>C(p.Q180P) variations. The c.396G>A(p.W132X) is a known mutation identified within more than 1 family, but c.539A>C(p.Q180P) is a novel variation. Her mother's DNA study showed she was a c.396G>A(p.W132X) heterozygote without c.539A>C(p.Q180P). Her younger sister's DNA study shows no variations. Her father refused to participate in the DNA study (Fig. 2).

Analysis of SLC22A5 identified the c.396G>A(p.W132X) and c.539A>C(p.Q180P) variations. The c.396G>A(p.W132X) is a known mutation but c.539A>C(p.Q180P) is a novel variation. Her mother’s DNA showed c.396G>A(p.W132X) heterozygote without c.539A>C(p.Q180P). Her younger sister’s DNA study shows no variations.
Follow-up of serum carnitine levels showed a low level even after administering L-carnitine solution. After changing medication to L-carnitine tablets 60mg/kg/day divided 3 times, serum carnitine level is improving (Table 1).

Follow-up of serum carnitine level
Discussion
During infancy and childhood, hypoglycemia occurs frequently in children with concomitant upper respiratory symptoms or acute gastroenteritis. Hypoglycemia develops when food intake is limited by anorexia and vomiting, and it typically occurs in the morning b efore breakfast. It most commonly occurs between 18 months and 5 years of age. Many hypoglycemic children are short and thin for their age and have decreased muscle mass due to the so-called ketotic hypoglycemia.
Other causes of hypoglycemia are hormonal abnormalities or metabolic defects. A first step to differential diagnosis is checking the level of urine or serum ketones. Then, the child is classified as either ketotic or hypoketotic.
In this patient, urine ketone was two positive although serum ketone was negative. Urine ketone can be false positive in cases of acidic urine, elevated urine specific gravity and some medications such as mesna. It should be reminded that ketonuria is different from ketonemia and it does not always indicate clinically ketonemia.
Hypoketotic hypoglycemia at presentation strongly suggests hyperinsulinemia or a defect in fatty acid oxidation. In this case she showed no ketonemia in her initial blood tests. We therefore conducted further evaluations as directed by many guidelines.
In the fasting state, fatty acid oxidation and ketone body formation is necessary for energy production1). Carnitine is essential for long-chain fatty acids transfer into mitochondria for fatty acid oxidation. Carnitine deficiency in children has been described in 2 disorders2). First, CDSP which is recessive defect of the muscle/kidney sodium-dependent plasma membrane carnitine symporter. It is expressed with cardiomyopathy or hypoketotic hypoglycemia in infancy. Second, a long-term oral medication of pivalate-conjugated antibiotics in which binding with carnitine can lead to carnitine deficiency. Secondary carnitine deficiency also can be seen in various organic acidemias and fatty acid oxidation defects.
CDSP is caused by deficiency of functional OCTN2 carnitine transporters3). This transporter is the most important factor managing body stores of carnitine. It is responsible for keeping the tissue gradient of carnitine in muscle and setting renal threshold for carnitine excretion. This carnitine transporter defect leads to decreased carnitine accumulation in tissue and increased losses of carnitine in the urine.
In this case, the child had recently taken antibiotics. Cefcapene Pivoxil HCl Hydrate (FLOMOX) is a third-generation cephalosporin that contains pivalic acid. Pivalic acid combines efficiently with carnitine to form pivalolycarnitine, which is excreted in urine. Long-term medication of pivalic acid containing antibiotics may leads to secondary carnitine deficiency in young children4). Ito et al. reported that this type of antibiotics may have adverse effects on mitochondrial function, even in short-term therapy5). In this case, her medication might have triggered her hypoglycemia and neurological symptoms6).
Clinical manifestations of CDPS can be a dominant metabolic or cardiac symptom with respect to age of onset7). The metabolic symptom presents usually before 2 years of age. It presents with mental changes and hypoketotic hypoglycemia when the child has an acute illness. In older children, cardiomyopathy sometimes associated with skeletal muscle weakness presents7). In this patient, no heart enlargement was detected on initial chest radiography and normal cardiac function on the follow-up echocardiography.
In this case, SLC22A5 gene analysis shows a novel variation on c.539A>C(p.Q180P) that is probably a novel mutation. Her younger sister shows no mutation. Further evaluations cannot be completed because of parental refusal to undergo genetic testing. Based in the gene analysis of case studies, there is no clear relationship between free carnitine level in plasma, OCTN2 genotype and disease severity at present8).
Newborn screening using tandem mass spectrometry can detect the level of acylcarnitines. A diagnosis of CDSP may be suspected early when tandem mass spectrometry can be done in the newborn period9,10). It should be considered both false-positive and false-negative results when the test is done too early due to the effect of maternal carnitine level. A repeat carnitine test must be done one week later to confirm the newborn’s carnitine disorder. In this patient, her mother remembered that newborn tandem mass spectrometry was unremarkable.
The treatment of CDSP is carnitine supplementation and responds well. The prognosis of disease depends on the severity of symptoms and onset age at diagnosis. Early detection and carnitine supplementation may prevent permanent sequels. It is reported that metabolic decompensation and skeletal and cardiac muscle function improves after carnitine supplementation11).
Long-term prognosis is favorable during carnitine supplementation is continued12). Although we supplemented our case with carnitine as an empirical treatment for seizures in the early treatment stages, generalized seizures progressed into status epilepticus. Her visual fixation and follow was disabled. The visual evoked potentials showed abnormal patterns and her visual acuity was seems to be impaired. The locomotion and transitional movements were also impaired and she needed rehabilitation therapy.
We report a case of systemic primary carnitine deficiency presented with hypoglycemic encephalopathy. It needs further evaluation tests when hypoketotic hypoglycemia is seen. The empirical carnitine supplementation may helpful for hypoketotic hypoglycemic seizures.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.