Transcriptional coordination of hepatic autophagy by nutrient-sensing nuclear receptor PPARα and FXR
Article information

Abstract
Nuclear receptors are in general ligand-dependent transcription factors that control a variety of mammalian physiologies including development, differentiation, proliferation, and homeostasis. Recent studies have found that two nutrient-sensing nuclear receptors, peroxisome proliferator-activated receptor α and farnesoid x receptor, responding to fasting or feeding state, respectively are able to regulate autophagy, an evolutionarily conserved catabolic process involved in lysosomal degradation. In this review, we discuss the role of these nutrient-sensing nuclear receptors in an aspect of transcriptional regulation of autophagy, and how these nuclear receptor-driven transcriptional programs integrate lipophagy, a lipid autophagy with fatty acid oxidation to coordinate hepatic lipid metabolism in the fasted state of the liver.
Introduction
The incidence of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) is a global pandemic particularly in developed countries probably due to western diets and sedentary lifestyle1). NAFLD and NASH also occur in children whose prevalence of prediabetes and type 2 diabetes is significantly increased2).
For the past 30 years, the diverse members of nuclear receptors (NRs) have a great impact on metabolic diseases345). Of particular, activation of nutrient-sensing NRs with their synthetic ligands has beneficial effects on the improvement of these pathogeneses678). However, a detailed understanding of molecular mechanisms by which controlling NRs treat metabolic disorders in liver and other tissues is not fully established. Here, we discuss a novel function of nutrient-sensing NRs, which exert the transcriptional control of autophagy, a self-eating process conserved in all eukaryotic cells. We first discuss a basic concept of NRs followed by the metabolic roles of 2 nutrient-sensing NRs peroxisome proliferator-activated receptor α (PPARα or NR1C1) and farnesoid x receptor (FXR or NR1H4). We then shift our focus to autophagy with an emphasis of macroautophagy. We finally discuss how nutrient-sensing NRs regulate macroautophagy, highlighting an integration of lipophagy, a lipid autophagy with fatty acid oxidation (FAO) to satisfy energy demands during starvation.
Overview of NR superfamily
As ligand-activated transcription factors (TFs), NRs serve as an interface between cellular or whole body environmental changes and our genome, providing an important link between transcriptional regulation and physiology. In particular, NRs play important functions in metazoan intercellular signaling because they converge diverse intra- and extracellular signals for initiating transcriptional programs for their relevant physiologies. Because of these unique roles in mammalian physiology, the activities of NRs are typically affected by a variety of environmental factors that function as ligands themselves or modulate ligand production5910). Although there are a few exceptions, NRs are usually composed of several domains: an N-terminal ligand-independent activation function 1 motif, a DNA-binding domain (DBD) with 2 zinc finger motifs, a flexible hinge domain, and a C-terminal ligand-binding domain (LBD) consisting of 12 α-helixes. Among them, the helix 12 corresponds to ligand-dependent activation function 2 motif, which undergoes a conformational change upon an agonist binding to the ligand-binding pocket of NR. This provides a docking site for coactivators, which lead to the recruitment of more transcriptional machinery proteins to initiate the transcription of a given NR target gene5). The human genome encodes 48 members of NRs that include classical endocrine receptors for steroid hormones, thyroid hormone, and fat soluble vitamins and their derivatives, adopted orphan receptors for fatty acids (FAs), phospholipids, cholesterol metabolites, and bile acids (BAs), and orphan receptors whose ligands have not been identified yet or may not exist at all5).
The comprehensive and regulatory functions of NRs on the target genes have led to intense research on these TFs, contributing to enhancing our understanding of complex molecular mechanisms of transcriptional biology. Therefore, NRs have a tremendous impact on almost every mammalian physiology at all levels of cells, tissues, organs and an entire organism. They are also tightly associated with many human diseases including cancer, cardiovascular diseases, innate and adaptive immune disorders, neurological diseases, and metabolic diseases1011). Moreover, NRs are often considered as valuable therapeutic targets to effectively treat human disease, which provides opportunities for developing better synthetic ligands with fewer side effects.
Nutrient-sensing NRs: PPARα & FXR
Among 48 members of NRs in human genome, PPARα and FXR are the most prominent NRs whose activities are regulated by the availability of nutrients or fasting-feeding cycle, which plays a critical role in maintaining energy homeostasis in metabolic tissues including the liver.
The finding that a synthetic chemical Wy-14,643 treatment to rodents dramatically increases the numbers of peroxisomes in the liver leads to the discovery of PPARα, a master TF for lipid metabolism under conditions of energy deprivation or fasting1112). Physiologic or pharmacologic activation of PPARα by fasting or synthetic ligands markedly promotes uptake, utilization, and breakdown of FAs via upregulation of genes involved in FA transport, FA binding, peroxisomal and mitochondrial β-oxidation, and ketogenesis91011). Moreover, PPARα also induces the expression of fibroblast growth factor 21 (Fgf21) gene, an endocrine and/or autocrine hormone with pleiotropic effects in many metabolic tissues, which show antiobesity and antidiabetic effects131415161718). As a ligand-activated NR, FAs such as arachidonic acid and polyunsaturated FAs, and C16:0-18:1 phosphatidylcholine have been reported to be endogenous agonist ligands for PPARα, which is also markedly activated by synthetic fibrate derivatives, a potent hypolipidemic drugs for patients with dyslipidemia61920). Fenofibrate, one of fibrate derivatives, effectively lowers serum triglycerides, which are applied to the treatment of coronary heart disease and NAFLD46). PPARα is highly expressed in tissues requiring effective FAO such as liver, brown adipose tissue, heart, skeletal muscle, kidney, intestine, and adrenal gland. Knockout studies of Pparα gene in mice also demonstrate that PPARα is absolutely required for proper lipid catabolism in the fasted state of the liver2122). PPARα forms heterodimeric complex with RXR, which typically binds to a direct repeat 1 response element (DR1 RE, 5' AGGTCA N AGGTCA 3', N; any single nucleotide) in the regulatory region of target genes1011).
FXR is a BA NR that controls BA homeostasis and its associated lipid and glucose metabolism, and that is required for normal liver regeneration and may mediate the beneficial effect of vertical sleeve gastrectomy in mice23242526). Chenodeoxycholic acid (CDCA), one of BA species has been found to be an endogenous agonist ligand for FXR in major metabolic tissues such as the liver and intestine27). It is generally believed that in these tissues FXR is activated by CDCA derived from an enterohepatic BA circulation process, suggesting that FXR potently responds to a fed state of the liver28). Therefore, FXR plays an essential role in BA homeostasis by remarkably suppressing hepatic expression of Cyp7a1 gene encoding cholesterol 7 α-hydroxylase, a rate-limiting enzyme of BA synthesis. FXR is also pharmacologically regulated by several synthetic or natural ligands such as obeticholic acid (OCA, also known as 6α-ethyl-CDCA or INT-747), GW4064, fexaramine, guggulsterone, and cafestol2930313233). Moreover, BA- or pharmacologic FXR activation in the intestine is sufficient for inducing the expression of fibroblast growth factor 15 (Fgf15) gene, an endocrine hormone participating in a part of the negative feedback regulations of BA synthesis, and in subsequent elevations of proteins and glycogen synthesis in the liver3435). As did PPARα, FXR also forms a heterodimeric receptor complex with RXR, which predominantly binds to an inverted repeat 1 response element (IR1 RE, 5' AGGTCA N TGACCT, N; any single nucleotide) in the regulatory region of many target genes. These transcriptional regulations of FXR target genes have a critical impact on the physiology of the fed state. Finally, US federal drug administration has recently approved the treatment of OCA, a semisynthetic CDCA analogue to patients with primary biliary cirrhosis, which might pave the way for diverse applications to other metabolic and/or inflammatory diseases3637).
General concept of autophagy
Autophagy, a self-eating process throughout all eukaryotic cells is a fundamentally conserved degradation mechanism involving lysosomal delivery of cargo molecules such as various soluble materials, membrane-enclosed organelles, and even invasive parasites3839). Although there is a basal autophagic activity in most cells and/or tissues, autophagy is highly inducible by numerous stimuli including physiologic stresses (e.g., nutrient deprivation, hypoxia, high temperature, high density condition, exercise, etc.), hormones (e.g., glucagon, etc.), pharmacologic reagents (e.g., rapamycin, Torin 1, etc.), or many disease conditions (e.g., cancer, myopathy, etc.)40). Although there are several types of autophagy depending on classifying criteria, macroautophagy is a major type of autophagy and have been most intensively studied so far compared to other types of autophagy. When the macroautophagy (herein referred to as autophagy) occurs, an isolated membrane called a phagophore sequesters cytoplasmic components to form double-membrane vesicle called an autophagosome (AG). AG then fuses with the lysosome to become an autolysosome (AL) where the autophagic cargos are degraded by acidic lysosomal hydrolases. It is also necessary to note that AL can be generated by a fusion of lysosome with amphisome, a vesicle created from a fusion between AG and late endosome (also called multivesicular body)41). Subsequently, biological building blocks (e.g., amino acids, FAs, nucleotides, and glucose) degraded from macromolecules or organelles within ALs are released into cytoplasm, which is recycled to synthesize new macromolecules or used for energy supplementations. In this view, autophagy itself acting as a cellular degradation process substantially links catabolism into anabolism3942). The catabolic function of autophagy is also associated with the quality control of many intracellular components by eliminating misfolded or unfolded proteins, or worn-out organelles. Therefore, cellular functions of autophagy are very diverse, ranging from eliminating superfluous organelles to providing amino acids and ATPs for energy supplementation and new protein synthesis to removing aggregate prone proteins for a quality control mechanism. Autophagy also plays an essential role in destroying invasive pathogens and subsequently presenting pathogen-derived antigens on the plasma membrane38). Finally too much autophagy seems to trigger certain types of cell death including apoptosis43). Beyond its cellular functions, autophagy has a broad impact on mammalian physiology and pathology including embryonic development, innate and adaptive immunity, neurodegenerative disease, cancer, heart disease and skeletal pathogenesis, ageing, metabolic disease, and so on44). On the contrary of a previous concept, suggesting that autophagy be a nonselective catabolic process, autophagy can degrade very selective cargo molecules and organelles. The latter includes proteins (aggrephagy), glycogen (glycophagy), lipid droplets (lipophagy), iron-bearing ferritins (ferritinophagy), the ribosome (ribophagy), the peroxisome (pexophagy), the endoplasmic reticulum (reticulophagy), the mitochondria (mitophagy), virus and bacteria (xenophagy), and so forth4546).
Transcriptional regulation of autophagy-related genes by PPARα & FXR
It has been relatively well known that acute regulation of autophagy by nutrient-sensing pathways is largely dependent on upstream signaling molecules including the mechanistic target of rapamycin complex 1 (mTORC1) and other kinases47). However, a longer-term regulation of autophagy has not been fully established yet. Particularly, transcriptional and epigenetic regulation of autophagy has been recently appreciated4849). Lee et al.50) and Seok et al.51) have shown that hepatic autophagy is controlled by several TFs including two nutrient-sensing NR PPARα and FXR, and cAMP responsive element binding protein (CREB), a well-known fasting activated TF for hepatic gluconeogenesis (Fig. 1). Both PPARα and CREB activated by fasting status induce hepatic autophagy via a direct upregulation of core autophagy-related genes, which also lead to lipophagy, one of selective autophagies, resulting in the release of free FAs from lipid droplets525354). It is of interest to note that PPARα is not only important for inducing FAO and ketogenesis to provide ATPs and alternative fuels for the brain, but also for increasing lipophagy to supply free FAs as substrates for the FAO during fasted state of the mammalian liver1119). It would be very intriguing to identify novel PPARα target genes encoding lipophagy adaptor proteins.
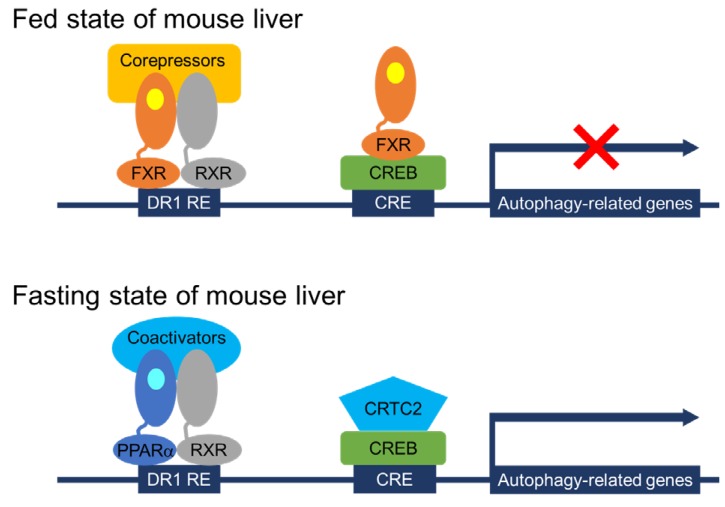
Working mechanisms of transcriptional regulation of autophagy by nutrient-sensing NRs and CREB. In the fed state of wild-type mice, FXR activated by its endogenous agonist CDCA (shown in yellow colored circle) binds to DR1 RE as a heterodimeric complex with RXR in the distal regulatory regions of autophagy-related genes. Moreover, activated FXR can also form a piggyback interaction with a CRE-bound CREB in the proximal regulator regions of autophagy-related genes via the dissociation of a coactivator CRTC2. These 2 different mechanisms may account for the transcriptional repression of autophagy-related genes by FXR activation in the fed state of mouse liver. In contrast, fasting-activated PPARα by its endogenous ligands, FFAs or PC (16:0-18:1)(shown in light blue colored circle), binds to DR1 RE via the formation of a heterodimeric complex with RXR in the distal regions of autophagy-related genes. CREB also recruits its coactivator CRTC2 in the proximal regions of autophagy-related genes. These complimentary transcription complexes ensure that fasting leads to turning on the expression of many core autophagy-related genes in the fasted state of mouse livers. Overall, activated PPARα or FXR competes with each other for binding to shared DNA sequences in the distal regions of autophagy-related genes. Additionally, FXR competes with CRTC2 for binding to CREB in the proximal regions of autophagy-related genes. NRs, nuclear receptors; CDCA, chenodeoxycholic acid; FFAs, free fatty acids; PC, phosphatidylcholine; FXR, farnesoid x receptor; RXR, retinoid x receptor; DR1 RE, direct repeat 1 response element; CRE, cAMP response element; CREB, cAMP response element binding protein; PPARα, peroxisome proliferator-activated receptor α; CRTC2, CREB regulated transcription coactivator 2.
In contrast, the BA receptor FXR represses hepatic autophagy by at least 2 mechanisms (Fig. 1). One mechanism proposed by Lee et al.50) is that FXR directly suppresses the expression of autophagy-related genes via a genomic competition with PPARα binding to DR1 RE that is often found in the regulatory regions of many autophagy-related genes. Consistent with this finding, previous studies have already demonstrated that FXR activation is able to repress ApoCIII and ApoA expression by binding to DR1 RE in the promoters of both genes where FXR may act as ligand-dependent transrepressor rather than transactivator5556). This mechanism is further supported by a comprehensive analysis of hepatic PPARα and FXR cistromes, showing their remarkable overlapping peaks containing DR1 RE throughout the whole mouse genome, which strongly suggests a possibility that both NRs compete for binding to shared sites in the regulatory regions of core autophagy-related genes, with opposite transcriptional outputs. On the other hand, the other mechanism proposed by Seok et al.51) is similar but distinct from that of Lee et al.50). They have shown that the piggyback binding of FXR to CREB without DNA binding in the promoter regions of autophagy-related genes is able to dissociate and subsequently expel CRTC2, a well-known coactivator from the nucleus. This leads to turning off the expression of many autophagy-related genes including TFEB, a master TF for the lysosomal biogenesis57). Previous studies by the Ballabio laboratory have shown that TFEB acts as a transcriptional activator for lysosomal and autophagy-related genes, which lead to the induction of autophagy. It is of interest to note that the catabolic effects of TFEB also depend on the induction of PGC1α and the presence of PPARα, suggesting a complementary function of TFEB-PGC1α-PPARα axis during fasting58). These results uncover complex but complementary genomic circuits in which transcriptional programs controlled by PPARα and CREB-CRTC2 induce hepatic autophagy, which is then markedly suppressed by FXR activation. Transcriptional circuits of autophagy-related genes governed by several TFs further integrate autophagy with long-term physiologic nutrient responses484959). These studies also suggest that beneficial effects of fibrate derivatives or OCA on patients with hyperlipidemia or primary biliary cirrhosis, respectively could be attributed to coordinating hepatic autophagy in addition to their known biochemical pathways, and that controlling autophagy by targeting PPARα and FXR might provide a novel therapeutic strategy for the pathogenesis of a wide range of human diseases.
Conclusions
It has been shown that PPARα and FXR, responding to fasting or feeding state, respectively are fundamental nutrient-sensing NRs, which orchestrate proper programs of transcription involved in FAO, ketogenesis, or BA homeostasis. The findings of Lee et al.50) and Seok et al.51) have extended the roles of these NRs to autophagy regulation. Although the details of underlying mechanisms by which PPARα and FXR counteract each other for autophagy regulation still need to be further elucidated, transcriptional regulation of autophagy by both NRs seems to be to some extent mTORC1-independent. It would also be of interest to identify direct PPARα or FXR target genes encoding key enzymes, which can modulate posttranslational modifications (PTMs) of core autophagy machinery proteins. Intriguingly, both mechanisms proposed by Lee et al.50) and Seok et al.51) are somewhat similar but still quite distinct from each other, suggesting that there should be complex transcriptional circuits governing mammalian autophagy, and that the expression of autophagy-related genes should be complementarily coordinated by several TFs including nutrient-sensing NRs, CREB, and TFEB. Taken together, nutrient deprivation allows mammalian cells to perform elaborate signaling pathways to rapidly trigger autophagy initiation by changing PTMs of autophagy machineries and to provide autophagy machinery proteins themselves by inducing the expression of autophagy-related genes. The latter may lead to the preparation of much longer-term of nutrient deprivation in the liver. Finally, investigations need to be performed to determine if the models proposed by Lee et al.50) and Seok et al.51) also control autophagy in other metabolic tissues, and if these mechanisms account for therapeutic effects of targeting PPARα or FXR on metabolic liver diseases.
Acknowledgments
We apologize for contributors whose work was not cited due to limited space. This research was supported by Kyungpook National University Research Fund, 2015.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.