Hypoparathyroidism, sensorineural deafness, and renal dysgenesis syndrome with a GATA3 mutation
Article information
Abstract
Hypoparathyroidism, sensorineural deafness, and renal dysgenesis syndrome is an autosomal dominant disease caused by mutations in the GATA3 gene on chromosome 10p15. We identified a patient diagnosed with hypoparathyroidism who also had a family history of hypoparathyroidism and sensorineural deafness, present in the father. The patient was subsequently diagnosed and found to be a heterozygote for an insertion mutation c.255_256ins4 (GTGC) in exon 2 of GATA3. His father was also confirmed to have the same mutation in GATA3.
Introduction
A syndrome with hypoparathyroidism, sensorineural deafness, and renal dysplasia was first reported by Barakat et al.1) in two brothers. It was named hypoparathyroidism, sensorineural deafness, and renal dysgenesis (HDR) syndrome by Hasegawa et al.2). This syndrome is inherited in an autosomal dominant fashion and is thought to be due to haploinsufficiency of GATA3. The known mutations of GATA3 include gross deletions, missense mutations, nonsense mutations, and small insertions or deletions that cause structural movement of the GATA3 gene3,4). In patients with HDR syndrome, hypoparathyroidism can be asymptomatic, but can also present as sensory problems, myalgia, and hypocalcemia severe enough to cause seizures. Sensorineural hearing loss is usually present from birth and is generally moderate to severe. It is most pronounced in bilateral and high frequency regions. Renal dysplasia may present as dysplasia, hypoplasia, aplasia, or vesicoureteral reflux. We identified a familial HDR syndrome case caused by a new mutation in GATA3, which we report here.
Case report
A 12-year-old Korean boy was admitted to Hallym University Kangdong Sacred Heart Hospital because of generalized tonic seizure with upward eyeball deviation for 7 minutes, half an hour before presenting to the hospital. This patient had no previous symptoms. He experienced fatigue 2 days before presentation, and the seizure that prompted his admission was his first. The patient was born by natural birth at 35 + 3 weeks and a weight of 2.5 kg. He was the first-born to nonconsanguineous parents. His father was diagnosed with hypoparathyroidism after having a seizure 4 years ago; he also had sensorineural hearing loss and was taking vitamin D and calcium carbonate tablets.
Physical examination findings included height 150 cm (25-50 percentile), weight 57.0 kg (75-90 percentile), blood pressure 100/60 mmHg, heart rate 80 bpm, respiratory rate 20/min, and temperature 36.9℃. The patient was healthy systemically but had a decreased level of consciousness. Head and neck examination did not reveal any signs of congenital malformations such as ocular hypertelorism. Chvostek's sign was negative, and no masses or lymph nodes were palpable in the neck. There was no neck stiffness or other abnormalities. On examination of the chest, breath sounds and heart sounds were normal. Abdominal examination revealed normal bowel sounds, without tenderness on palpation or rebound tenderness. There was no edema in the four limbs, costovertebral pressure pain, or rashes on the skin.
At the time of hospitalization, laboratory finding showed the following results: total calcium 6.2 mg/dL (reference range [RR], 8.8-10.8 mg/dL), phosphorus 7.1 mg/dL (RR, 3.8-6.5 mg/dL), ionized calcium 0.54 mmol/L (RR, 0.95-1.5 mmol/L), magnesium 2.2 mg/dL (RR, 1.6-2.6 mg/dL), serum alkaline phosphatase 223 IU/L (RR, 60-360 IU/L). The level of 25(OH)-vitamin D was 14.90 ng/mL (RR, 8.0-51.9 ng/mL), 1,25(OH)2vitD 61.9 pg/mL (RR, 19.6-54.3 pg/mL), parathyroid hormone 11.76 pg/mL (RR, 15-65 pg/mL), and calcitonin 2.93 pg/mL (RR, 0-10.0 pg/mL). Urinalysis showed no proteinuria or hematuria. Urine microscopy revealed 1-4/mm3 white cells and 1-4/mm3 red cells, with no significant abnormalities. Head and neck computed tomography (CT) showed basal ganglia calcification.
Since the father had hearing loss and hypoparathyroidism, HDR syndrome was considered in the patient, who underwent a hearing test and abdominal ultrasonography. On tympanometry, there was no hearing function in the left ear, while the right ear was normal. Pure tone audiometry revealed sensorineural hearing loss in both ears (Fig. 1). Abdominal ultrasound showed absence of the renal fossa in the right kidney, indicating dysplasia of the kidney. GATA3 gene analysis showed that the patient and his father were heterozygotes with a mutation in exon 2: an insertion of 255_256ins4 (GTGC) (Fig. 2). After hospitalization and treatment with intravenous calcium and vitamin D, the patient's blood tests showed improvement, with a total calcium level of 8.7 mg/dL, phosphorus 8.1 mg/dL, and ionized calcium 1.02 mmol/L. From day 2 of hospitalization, the patient was started on oral calcium 50 mg/kg/day, and 1,25(OH)2vitD 0.5 µg/day and was subsequently discharged with outpatient follow-up. The patient presently has normal levels of calcium and phosphorus. His father has also been on treatment and maintained normal levels of calcium and phosphorus.
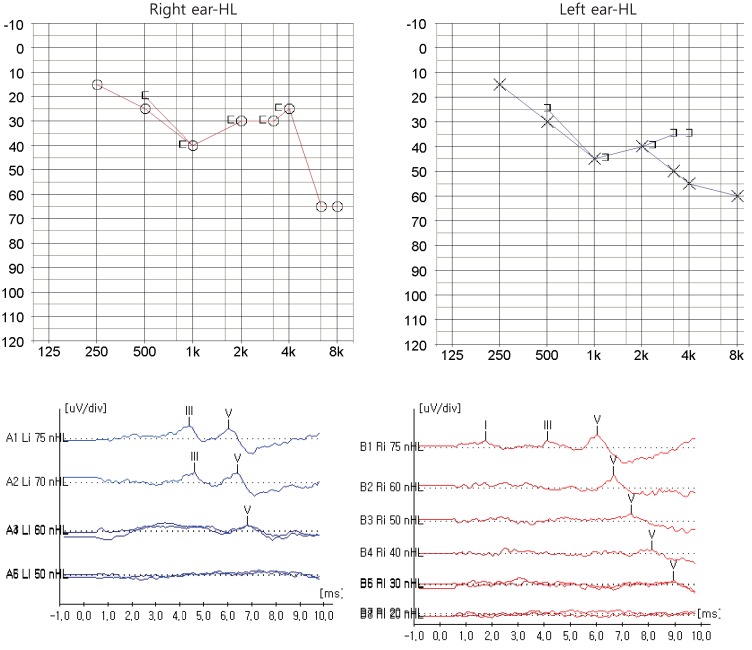
Pure tone audiometry and auditory evoked potential show sensorineural hearing loss (HL) both ear (especially left ear, higher frequency) at diagnosis.
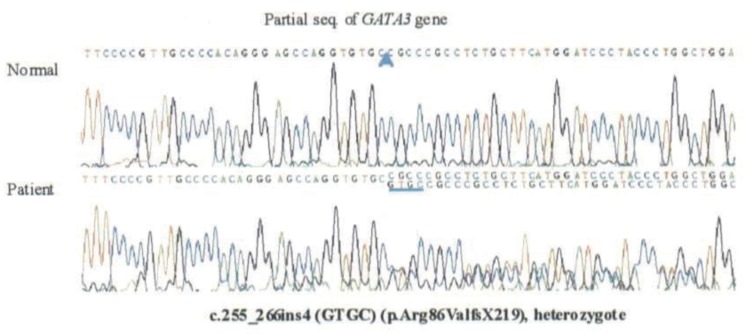
A mutation was confirmed in exon 2 of the GATA3 gene (c.255_266ins4 (GTGC)) and was identified in both our patient and his father.
Discussion
Bilous et al.5) first established in 1992 that HDR syndrome was an autosomal dominant inherited disease. Van Esch et al.3) subsequently showed that HDR syndrome was primarily caused by haploinsufficiency of GATA3 on chromosome 10p15. The GATA3 gene is 20 kb long, containing six exons6), and encodes a 444-amino acid transcription factor comprising two transactivation domains (TA1, TA2) and two zinc finger domains (ZF1, ZF2)7). GATA3 belongs to a family of zinc finger transcription factors (GATA family) that are involved in vertebrate embryonic development8). GATA family was called because the GATA transcription factor binds to (A/T) GATA (A/G). So far, six GATA genes have been identified in mammals that play a significant role in the genetous transduction chain reaction9). GATA3 expression products can be found in various organs from 4 weeks after birth10), and the translation products of GATA3 are consistently found in the kidneys, otic vesicles, and parathyroid gland, all of which are involved in HDR syndrome11). Mutations that can cause HDR syndrome include gross deletions, missense mutations, nonsense mutations, and small insertions or deletions, which cause structural movement of the GATA3 gene. In our patient and his father, a mutation (insertion at exon 2 of GATA3, c.255_256ins4 (GTGC)) was found. DNA sequencing revealed a insertion of the GTGC nucleotide at position 255_256, which results in a frameshift beginning at codon 86 and leading to a premature stop signal at codon 304 (p.Arg86ValfsX219).
Hypoparathyroidism in HDR syndrome can range from asymptomatic to myalgia, sensory problems, and a pronounced tetany pattern caused by hypocalcemia. The concentrations of parathyroid hormone range from low to high even in some individuals12). The finding of an active increase in cyclic adenosine monophosphate (cAMP) levels in patients who receive parathyroid hormone indicates normal affinity of the parathyroid hormone receptor3).
Early-onset sensorineural hearing loss is seen in most patients with HDR syndrome, and these patients may be diagnosed with HDR syndrome after initially being identified with hearing problems13). The hearing loss is characterized by a biased hearing sound range. Patients have limited voice recognition at low amplitudes, and the decrease in the extent of hearing loss is moderately severe for moderate sounds. The hearing loss progressively worsens with age, becoming more severe at high frequencies. Although patients are not able to evoke otoacoustic emissions, they have normal auditory brainstem response between peaks, suggesting that the mechanism of hearing loss involves the outer hair cells14). Our patient had pronounced hearing loss at high frequencies, suggesting sensorineural hearing loss, and hence it is likely that his hearing loss will worsen with age. Therefore, regular examinations of hearing function and identification of other factors that can exacerbate hearing loss should be performed for prevention. The patient in this report is currently under regular follow-up by otolaryngologists.
The kidney symptoms of HDR syndrome can present bilaterally or unilaterally and can involve both developmental abnormalities such as renal dysplasia, including dysplasia, hypoplasia, aplasia, and vesicoureteral reflux. Also renal disorders include functional abnormalities such as proteinuria, hematuria, renal tubular acidosis, nephrocalcinosis15) and renal failure16). It has been reported that even among family members carrying the same GATA3 mutation, the penetration of kidney disease and parathyroid gland disease can vary. Hernaderz et al.12) reported that a daughter had unilateral renal agenesis and her mother had a nonfictional kidney a patient although they were diagnosed same GATA3 mutation. In our cases, the father did not have kidney disease, but the patient had the classical triad HDR syndrome. Family members sharing the same mutations can express different kidney abnormalities2,5). Our patient had hypoplasia of the right kidney, but urinalysis did not show proteinuria, hematuria, or hypercalciuria.
Other manifestations such as pyloric stenosis4) and abnormalities of the uterus or vagina leading to dysmenorrhea, menorrhagia, or amenorrhea have been reported12). Those with large chromosomal deletions can present with body part malformations, cataracts, basal ganglia calcifications, severe mental weakness, or autism17), suggesting that the chromosomal deletion may involve other gene(s). Those with point mutations displayed goiter18), unilateral megaencephaly with seizures19), familial urological abnormalities12), bilateral cataracts, basal ganglia calcification, and psoriasis20). Our patients showed basal ganglia calcification on brain CT.
In summar y the authors identified a familial HDR syndrome case caused by a mutation in GATA3. HDR syndrome is an autosomal dominant disease that involves hypoparathyroidism, sensorineural hearing loss, and kidney abnormalities. It is caused by mutations in the GATA3 gene leading to its haploinsufficiency. Patients who present with hypoparathyroidism, or hearing loss, or kidney abnormalities with a history of one or more of these symptoms should be suspected for HDR syndrome. The patients and their family members can be diagnosed using GATA3 gene analysis. However, our understanding of HDR syndrome to date is not complete, and future research is needed on this disease.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.