Clinical, endocrinological, and molecular characterization of Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism: a single center experience
Article information
Abstract
Purpose
Isolated gonadotropin-releasing hormone (GnRH) deficiency (IGD) is classified as Kallmann syndrome (KS) with anosmia and normosmic idiopathic hypogonadotropic hypogonadism (nIHH). This study was undertaken to investigate the clinical, endocrinological, and molecular characteristics in Korean patients with KS and nIHH.
Methods
Twenty-six patients from 25 unrelated families were included. Their clinical, endocrinological, and radiological findings were analyzed retrospectively. Mutation analysis of the GNRH1, GNRHR, KISS1, KISS1R, PROK2, PROKR2, TAC3, TACR3, FGF8, FGFR1, and KAL1 genes was performed in all patients. CHD7 and SOX10 were analyzed in patients with CHARGE (Coloboma, Heart defects, choanae Atresia, Growth retardation, Genitourinary abnormality, Ear abnormality) features or deafness.
Results
Of the 26 patients, 16 had KS and 10 had nIHH. At diagnosis, mean chronologic age was 18.1 years in males and 18.0 years in females; height SDS were -0.67±1.35 in males, -1.12±1.86 in females; testis volume was 2.0±1.3 mL; and Tanner stage was 1.5. There were associated anomalies in some of the KS patients: hearing loss (n=6) and congenital heart disease (n=4). Absence or hypoplasia of the olfactory bulb/sulci was found in 84.62% of patients with KS. Molecular defects in KAL1, SOX10, and CHD7 were identified in 5 patients from 4 families (16.0%, 4/25 pedigrees). After sex hormone replacement therapy, there were improvement in sexual characteristics and the sexual function.
Conclusion
This study described the clinical, endocrinological, and molecular genetic features in IGD patients in Korea. Although the mutation screening was performed in 10 genes that cause IGD, molecular defects were identified in relatively small proportions of the cohort.
Introduction
The hypothalamic-pituitary-gonadal (HPG) axis plays a crucial role in the development and progression of puberty. The pulsatile secretion of gonadotropin-releasing hormone (GnRH) into the hypophyseal-portal vessels controls the synthesis and release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in the anterior pituitary gland, which then stimulates the gonads to produce sex steroids and gametes1).
Isolated GnRH deficiency (IGD) is a rare disorder with an estimated incidence of one case per 48,000 births2). It is caused by a defect in the HPG axis resulting in low levels of sex steroids and delayed or absent puberty. IGD is divided into anosmic hypogonadotropic hypogonadism (Kallmann syndrome, KS) and normosmic idiopathic hypogonadotropic hypogonadism (nIHH)3). About 60% of patients with IGD present with anosmia or hyposmia (KS) by total or partial defects of the olfactory bulb due to the fact that the GnRH neurons are primarily derived from progenitor cells in the nasal compartment and migrate along the fibers derived from the olfactory system across the cribriform plate to the forebrain1,4,5). Associated non-reproductive phenotypes include cleft lip/palate, hearing impairment, renal agenesis, and mirror movement6). The remaining 40% of patients with IGD have a normal sense of smell (nIHH)1).
Mutations in at least 19 genes contribute to the molecular basis of nIHH/KS. The genetic defects are classified according to the pathophysiology: defects in the neurodevelopmental pathway (KAL1, NELF, SOX10), neuroendocrine pathway (GNRH1, GNRHR, KISS1, KISS1R, TACR3, TAC3), and both (PROKR2, PROK2, FGFR1, FGF8, CHD7, HS6ST1)1,7,8,9,10,11,12). KS is genetically heterogeneous, and most KS cases occur in a sporadic form. In familial KS, autosomal recessive, autosomal dominant, and X-chromosomal recessive inheritance has been described11). However, these genes only account for half of the etiologies of all nIHH/KS patients12).
Because of the rarity of IGD and its under-recognition by physicians, few cases have been reported in Korea. Therefore, this study described the clinical, endocrinological, and molecular characteristics in patients with IGD in Korea.
Materials and methods
1. Clinical characteristics
This study included 26 patients from 25 unrelated families diagnosed with IGD. Criteria for diagnosis of IGD included: (1) absent or delayed puberty: testis volume<4 mL at 15 years in males, absent of thelarche at 13 years in females; (2) clinical signs or symptoms of hypogonadism (micropenis, cryptorchidism, no breast budding, etc.); (3) serum testosterone level below 1 ng/mL in males or serum estradiol level below 20 pg/mL in females in the presence of low or normal gonadotropins; and (4) otherwise normal anterior pituitary function13).
The following clinical parameters were obtained by retrospective review of the patients' medical records: history of cryptorchidism, micropenis, dental agenesis, cleft lip or palate, prior pubertal development, prior sex hormone treatment, associated nonreproductive phenotypes, sense of smell, and family history. All patients underwent a complete physical examination to determine height, body weight, and associated anomalies, and synkinesis. In the male patients, testicular volume was measured using the Prader orchidometer. In the female patients, the presence of breast buds and menstruation was checked. Pubertal development was rated according to the guidelines recommended by Marshall and Tanner14). Micropenis was defined by a stretched penile length of less than 30 mm15,16). Bone age was determined by the Greulich and Pyle method using x-rays of the hand and wrist17).
Of the 26 IGD patients, 24 self-reported their olfactory function and 2 patients undertook the 16 item smell test (Korean Version of Sniffin' Sticks Test, KVSS test II). The KVSS identification test using 16 odorants familiar to Koreans was used to evaluate olfactory function. The T.D.I. score (olfactory Threshold, odor Discrimination, odor Identification score) was used to differentiate the cases into normosmia (30.73±4.75), hyposmia (22.82±6.81), and anosmia (10.69±3.37)18).
Renal structures were assessed by abdominal ultrasound. Pelvic ultrasound was also performed in females to determine the size and shape of the uterus. Brain magnetic resonance imaging (MRI) was used to visualize the olfactory bulbs, sulci, and inner ear structures.
2. Endocrine evaluations
The GnRH stimulation test was performed in all patients. Before GnRH injection, the basal hormone levels of LH, FSH, and testosterone (male) or estradiol (female) were measured. After injection of Relefact LHRH 60 µg/m2 (maximum dose, 100 µg) the levels of LH and FSH at 15, 30, 60, and 90 minutes were measured. The estradiol or testosterone levels were measured by radioimmunoassay (RIA; Coat-A-Count, Diagnostic Products, Los Angeles, CA, USA). The LH and FSH levels were determined by immunoradiometric assay (IRMA-mat, Byk-Sangtec Diagnostica, Hessen, Germany).
3. Molecular genetic analysis
After obtaining informed consent, genomic DNA was extracted from peripheral blood lymphocytes using a Gentra Puregene Blood Kit according to the manufacturer's protocol (QIAGEN, Hilden, Germany). In all the patients, all the exons and intronic flanking regions of the GNRH1 (MIM 152760), GNRHR (MIM 138850), KISS1 (MIM 603286), KISS1R (MIM 604161), PROK2 (MIM 607002), PROKR2 (MIM 607212), TAC3 (MIM 162330), TACR3 (MIM 162332), FGF8 (MIM 600483), FGFR1 (MIM 136350), and KAL1 (MIM 308700) genes were amplified by polymerase chain reaction with specific primers and then directly sequenced as previously described19). Mutation analyses for the CHD7 (MIM 608765) and SOX10 (MIM 602229) genes were performed selectively in patients with Coloboma, Heart defects, choanae Atresia, Growth retardation, Genitourinary abnormality, Ear abnormality (CHARGE) features CHARGE (Coloboma, Heart defects, choanae Atresia, Growth retardation, Genitourinary abnormality, Ear abnormality) and deafness, respectively20,21). Chromosomal analysis was performed in 24 patients (15 with KS and 9 with nIHH).
Results
1. Clinical characteristics
Of the 26 patients, 16 (61.5%) were KS and 10 (38.5%) were nIHH. The KS group included 14 males and 2 females, and the nIHH group included 8 males and 2 females. The mean age at diagnosis was 18.05±3.85 years (range, 15-31 years) in males, 18.00±3.92 years (range, 14-23 years) in females. At diagnosis, the difference between bone age and chronologic age (BA-CA) was -2.03±1.38 years (range, 0-4.0 years) in males, -3.75±3.5 years (range, 0-8.0 years) in females and the mean height z-score was -0.67±1.35 (range, -3.14 to 1.39) in males, -1.12±1.86 (range, -2.88 to 1.50) in females. In males, mean testis volume was 2.0±1.3 mL (range, 0.5-3 mL); stretched penile length was 3.8±1.4 cm (range, 1-7 cm). Mean pubic Tanner stage was 1.5 (range, 1-3).
Most patients with KS manifested cryptorchidism (n=2), sensorineural hearing loss (n=6) and congenital heart disease (n=4, pulmonary artery hypoplasia, ventricular septal defect, aortic valve stenosis). Of the 16 patients with KS, one patient had Waardenburg syndrome and two had CHARGE syndrome. The other associated phenotypes were ptosis (n=1), scoliosis (n=2), osteoporosis (n=1), alopecia (n=1), and obesity (n=7). There were no patients showing synkinesis or renal agenesis.
Of the 10 patients with nIHH, the associated features were cryptorchidism (n=2), osteoporosis (n=2), obesity (n=2), vesicoureteral reflux (n=1), ptosis (n=1), syndactyly (n=1), and scoliosis (n=1) (Table 1).

Associated congenital abnormalities of patients with KS and nIHH
Brain MRI was performed in 22 patients (13 with KS, 9 with nIHH) to detect the presence of an abnormal olfactory structure or other brain lesion. Absence or hypoplasia of olfactory bulb/sulci was found in 11 of those 13 KS patients (84.62%) (Fig. 1). No abnormal brain MRI finding was observed in the 9 patients with nIHH.
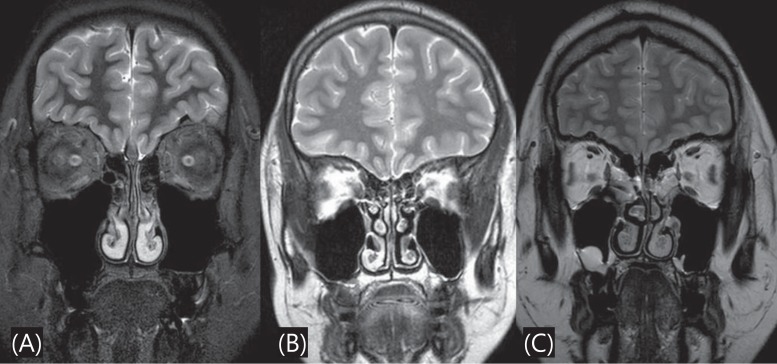
Brain magnetic resonance imaging of the olfactory bulb in a patients with Kallmann syndrome. (A) T2-weighted sequence in coronal plane shows very small olfactory bulb, (B) Fluid-attenuated inversion recovery (FLAIR) sequence shows no olfactory bulb, and (C) normal olfactory bulb and sulci.
2. Endocrinologic profiles
The GnRH stimulation test was performed at mean age 18.05±3.85 years in males, 18.00±3.92 years in females. The baseline LH and FSH levels were 0.96 mIU/mL (range, 0.2-2.7 mIU/mL) and 0.97 mIU/mL (range. 0.2-2.8 mIU/mL), respectively. Peak LH and FSH were 3.84±3.54 (range, 1.0-16.4 mIU/mL) and 3.65±1.79 mIU/mL (range, 1.6-8.1 mIU/mL), respectively (Table 2). The results suggest prepubertal patterns of gonadotropin secretion by GnRH stimulation. The average testosterone level was 0.46 ng/mL (range, 0.04-0.85 ng/mL) in males and the average estradiol level was 11.93 pg/mL in females (range, 10-13.1 pg/mL). The levels of testosterone and estradiol were lower than the age and sex-matched reference range.

Endocrinological features of patients with KS and nIHH
3. Molecular genetic analysis
Of the 26 patients from 25 unrelated families, molecular defects were identified in 5 patients from 4 families (16%, 4/25 pedigrees) in KAL1, SOX10, and CHD7. Karyotype was normal in 24 patients who underwent chromosome analysis. A novel heterozygous p.W380X mutation in KAL1 was identified in two male siblings from a nonconsanguineous marriage. They presented with anosmia/hyposmia and delayed puberty at age 14 and 16 years, respectively. The patient with a heterozygous p.M112I mutation in SOX10 presented with hypogonadotropic hypogonadism, anosmia, and deafness at age 31. The SOX10 p.M112I mutation was previously reported in Waardenburg syndrome22). Two unrelated female patients with typical and partial/incomplete CHARGE syndrome, as determined by Verloes' criteria23), were heterozygous for frameshift (p.A608Gfs*4) and nonsense (p.W983X) mutations in CHD7, respectively. Both mutations are not reported elsewhere, and each patient has been treated with estrogen/progesterone cyclic therapy.
4. Clinical course and outcomes
All twenty-six patients (21 males, 4 females) received sex hormone replacement therapy. One patient was treated in another hospital due to relation to hometown. Twenty-one males were given testosterone enanthate (50-100 mg, intramuscularly every 4 weeks) then the dose was increased to 200-250 mg. Conjugated estrogen was administered to 4 females at a dose of 0.3 mg per day and the dose was subsequently increased up to 1.25 mg/day. The mean follow-up period was 50.6 months (range, 3-108 months). During the follow-up period, 25 patients reached the final adult height. In males, 21 patients reached the final adult height with the mean height of 173.96±7.08 cm (0.27±1.11 standard deviation score [SDS]; range, 158-185 cm) that was similar to their midparental height of 171.29±4.10 cm (-0.33±0.75 SDS). In females, 4 patients reached the final adult height with the mean height of 157.85±9.96 cm (0.97±0.82 SDS; range, 147.6-171.5 cm) that was similar to their midparental height of 158.67±4.37 cm (-0.42±0.87 SDS). In males, there were significant improvement in stretched penile length (3.6 cm vs. 6.61 cm, P<0.001), pubic hair (Tanner stage 1.33 vs. 3.71, P<0.001), and laryngeal prominence before and after treatment. However, there were no significant differences in testicular volume (Table 3). During the treatment period, a few side effects of androgen, such as acne (n=4) and erythrocytosis (n=1), occurred in 5 patients. The prostate specific antigen (PSA) levels were normal. In females, there was significant changed pubic hair (Tanner stage 1.25 vs. 1.50, P<0.001) (Table 4).

Clinical outcome of 21 male patients with KS and nIHH after hormonal replacement

Clinical outcome of 4 female patients with KS and nIHH after hormonal replacement
Discussion
This study described the clinical and endocrinological characteristics, and the molecular genetic spectrum of IGD in a relatively large cohort in a Korean population. Because mutation screening was only performed in 11 genes (KAL1, FGF8, FGFR1, KISS1, KISS1R, PROK2, PROKR2, GNRHR, TAC3, and TACR3) known to cause IGD, molecular defects were identified in relatively small proportions of the cohort (16%; 4/25 unrelated families). IGD is one of the most common causes of pubertal delay and it is caused by either congenital defects of the HPG axis or acquired problems8). Acquired hypogonadotropic hypogonadism can be caused by drugs, infiltrative or infectious lesions, prolactinoma, radiation to the hypothalamus/pituitary gland, and system diseases such as hemochromatosis, sarcoidosis, and histiocytosis X13,24).
The pathogenetic mechanism of congenital IGD involves the failure of GnRH neurons in the hypothalamus to differentiate or develop, resulting in the lack of GnRH secretion or apulsatile GnRH secretion25). Recently, the molecular mechanisms that regulate the activation of GnRH have been determined and other abnormal phenotypes that are commonly associated with hypogonadism have also helped to identify the underlying molecular defects26). The most commonly associated phenotype is anosmia that is caused by impaired migration of GnRH and olfactory neurons. Similar to the development of olfactory fibers, GnRH neurons are formed in the nasal placode (olfactory epithelium) and migrate through the olfactory pathway to the hypothalamus. KS is genetically heterogeneous and most cases present as sporadic. Many of the gene mutations associated KS have been identified in KAL1 and FGFR1 (KAL2) accounting for about 15% of KS1). The KAL1 gene was mapped to chromosome Xp22.32 and it consists of 14 exons. Most KAL1 mutations are nucleotide insertions or deletions resulting in frameshift mutations or premature stop codons24). The monoallelic mutation in FGFR1 is found in approximately 10% of KS cases27). In addition, mutations in CHD7 have also been shown to cause KS21). Mutations in KISS1, TAC3 and TACR3 were identified in nIHH1,7). One study has reported that mutations were identified in five genes (GNRHR, TACR3, TAC3, KISS1R, and KISS1) in 77% of families with autosomal recessively inherited nIHH28).
KS is associated with many additional abnormalities. The most commonly described abnormalities include: reproductive phenotypes (micropenis, cryptorchidism) and nonreproductive phenotypes (synkinesis, hearing loss, renal anomalies, cleft lip/palate, and dental agenesis). Less frequent phenotypes are musculoskeletal anomalies, such as clinodactyly and camptodactyly; oculomotor anomalies, such as ptosis and nystagmus; and cardiac defects. Cardiac defects include atrial septal defect, ventricular septal defect, right-sided aortic arch, double-outlet right ventricle, transposition of the great arteries, and arrhythmias29). Micropenis was noted in up to 65% of males with KS. Cryptorchidism has been reported in up to 73% of males with KS30). Cleft lip/palate was found in up to 13%-14% of individuals with KS31). The incidence of hearing loss in patients with KS has been reported to be as high as 28% in previous case series32).
In nIHH, the prevalence of cryptorchidism has been known to be lower than it is in KS33). However, in our study, 8 males with nIHH were found to have micropenis (n=4, 50%) or cryptorchidism (n=2, 25%), so there is no significant difference in the prevalence of cryptorchidism between KS and nIHH (Table 1). In this study hearing defect was much more preponderantly observed in KS than in nIHH. There is no sensorineural hearing loss or congenital heart defects in nIHH. In addition, no patient presented with cleft lip/palate, synkinesis, or renal agenesis in KS. As many of these anomalies are identifiable at birth, recognition of these phenotypes allows for the opportunity for early diagnosis4).
Brain MRI is helpful for confirmation of diagnosis and it is the modality of choice in assessing the olfactory tracts and sulci. A coronal view is the best suited plane for overview of the olfactory tract34). Olfactory bulbs are normally seen as well-defined structures along the cribriform plate. Olfactory sulci are seen between the gyrus rectus and the medial orbital gyrus35,36). Koenigkam-Santos et al.37) found that olfactory bulb and sulcus aplasia were the most common findings in KS patients. In this study, absence or hypoplasia of olfactory bulb/sulci was found in 11 of 13 KS patients (84.62%).
Three treatment options are available for KS and nIHH: gonadal steroid hormones, human gonadotropins, and pulsatile GnRH. Androgen therapy could induce only virilization38,39). Pulsatile GnRH treatment has been known to have the advantage of testicular growth, fertility, virilization in men and follicular development and ovulation in women. In that therapeutic approach, a portable infusion pump administers GnRH in a pulsatile manner every 2 hours with doses ranging from 100 ng/kg to 400 ng/kg40). Recently, treatment with human chorionic gonadotropin (hCG) alone or in combination with recombinant human FSH or human menopausal gonadotropins (hMG) was introduced to induce sperm production. The gonadotropin treatment starts with the administration of 1,000 IU to 2,500 IU of hCG twice a week for 8 to 12 weeks. In individuals who do not have sufficient endogenous FSH, treatment can continue with the coadministration of 75 IU to 150 IU of hMG three times per week for up to 18 months. This combined treatment provides considerable testicular growth in most patients and spermatogenesis occurs in up to 90% of patients40).
In the present study, of the 26 patients, 25 (21 males, 4 females) received sex hormone replacement therapy. The side effects of testosterone replacement therapy are prostate hyperplasia, prostate cancer, sleep apnea, erythrocytosis, and abnormal liver function test. For prostate cancer, a digital rectal examination and measurement of serum PSA were performed three to six months after initiation of treatment, and then once a year. The secondary sexual characteristics and the sexual function of the patients who received hormone replacement therapy were markedly improved after treatment. There were significant differences in penile length and pubic hair before and after treatment in males (Table 3). In this study, exogenous estrogens were used to initiate secondary sexual development in prepubertal females (Table 4). Combined therapy with progestin is eventually needed to prevent endometrial hyperplasia.
In conclusion, the result of this study further enhances our understanding of the clinical, hormonal, and molecular genetic characteristics of KS and nIHH and the effects of sex hormone replacement therapy. Early diagnosis will aid in appropriate patient care and more accurate recurrence risk counseling. Genetic analysis of the IGD-causing genes is needed to verify the molecular genetic causes of KS and nIHH.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.