Gitelman syndrome combined with complete growth hormone deficiency
Article information
Abstract
Gitelman syndrome is a rare autosomal recessive hereditary salt-losing tubulopathy, that manifests as hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria. It is caused by mutations in the solute carrier family 12(sodium/chloride transporters), member 3 (SLC12A3) gene encoding the thiazide-sensitive sodium chloride cotransporter channel (NCCT) in the distal convoluted tubule of the kidney. It is associated with muscle weakness, cramps, tetany, vomiting, diarrhea, abdominal pain, and growth retardation. The incidence of growth retardation, the exact cause of which is unknown, is lower than that of Bartter syndrome. Herein, we discuss the case of an overweight 12.9-year-old girl of short stature presenting with hypokalemic metabolic alkalosis. The patient, on the basis of detection of a heterozygous mutation in the SLC12A3 gene and poor growth hormone (GH) responses in two provocative tests, was diagnosed with Gitelman syndrome combined with complete GH deficiency. GH treatment accompanied by magnesium oxide and potassium replacement was associated with a good clinical response.
Introduction
Gitelman syndrome is a rare autosomal recessive renal tubular disorder1). Characteristic laboratory abnormalities are hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria. It is usually caused by mutations in the SLC12A3 gene encoding the thiazide-sensitive sodium chloride cotransporter channel (NCCT) in the distal convoluted tubule of the kidney2). Diagnosis is most often made incidentally during adolescence or early adulthood. Although Gitelman syndrome is sometimes asymptomatic, it can present with clinical symptoms such as fatigue, muscle weakness, cramps, tetany, vomiting, diarrhea, and abdominal pain. Growth retardation and poor weight gain are common features, but are less severe than in Bartter syndrome3). Although potassium depletion may have some negative effect on growth, the exact cause of growth retardation in Gitelman syndrome is unknown. Reports of Gitelman syndrome combined with growth hormone (GH) deficiency are quite rare. Here, we report on a girl with Gitelman syndrome, diagnosed by molecular evaluation, combined with GH deficiency.
Case report
A 12.9-year-old girl visited our department of pediatrics complaining of chronic abdominal pain and diarrhea. She had also experienced tetany of the hand and foot for several months. At her full-term birth, her weight was 2.75 kg. Her family history had no history of renal, endocrinologic, or cardiovascular disease. Her father's height was 170 cm, her mother's 155 cm, and the midparental height, 156 cm. She had no history of diuretics use. Her height was 138.4 cm (<3rd percentile, -2.33 standard deviation score [SDS]); her weight was 43.3 kg (25th-50th percentile, -0.23 SDS); her body mass index was 22.7 kg/m2 (85th-90th percentile). The puberal stage was breast II and pubic hair I, and her bone age, according to the Greulich-Pyle method, was 11 years. Her blood pressure was 100/60 mmHg. Initial biochemical analysis revealed, on the basis of the following values, metabolic alkalosis with hypokalemia: sodium 139 mEq/L, potassium 2.4 mEq/L, chloride 98 mEq/L, blood urea nitrogen 5.8 mg/dL, creatinine 0.4 mg/dL, aspartate aminotransferase 48 U/L, alanine aminotransferase 67 U/L, pH 7.467, PCO2 47.4 mmHg, HCO3- 34.5 mmol/L (normal range, 22 to 26 mmol/L), base excess 9.6 mmol/L (-4 to 2 mmol/L), total calcium 9.9 mg/dL, phosphorus 4.7 mg/dL. Further laboratory investigation showed the following: magnesium 1.48 mg/dL (1.5 to 2.3 mg/dL), serum osmolality 218 mOsmol/kg (275 to 295 mOsmol/kg), renin 32.66 ng/mL/hr (<4.2 ng/mL/hr), and aldosterone 361.28 pg/mL (20 to 220pg/mL). Urine test results were as follows: Na+ 61 mEq/L, K+ 29.0 mEq/L, Cl- 57 mEq/L, osmolality 641 mOsmol/kg (50 to 1400 mOsmol/kg), spot urine calcium/creatinine ratio <0.02 mg/mg (0.03 to 0.2 mg/mg), 24 hours urine calcium 0.62 mg/kg (1.0 to 4.0 mg/kg/day), 24 hours urine magnesium 0.74 mg/kg (2.82±0.79 mg/kg/day). The calculated transtubular potassium gradient (TTKG) had increased to 8.078.
The patient had normal thyroid and parathyroid function. Abdominal ultrasound showed normal kidneys and diffuse mild fatty infiltration of the liver. GH stimulation tests for short stature revealed complete deficiency (Table 1). Magnetic resonance imaging of the brain revealed no remarkable findings. Her intelligence quotient as measured by the Korean Wechsler Intelligence Scale for Children III test was 67. Whole-blood DNA sequencing showed heterozygous mutation in the SLC12A3 gene (Fig. 1). Although her sister and brother were not genetically evaluated, they showed normal electrolyte and blood gas readings.

GnRH and GH stimulation tests showed normal pubertal response and complete GH deficiency
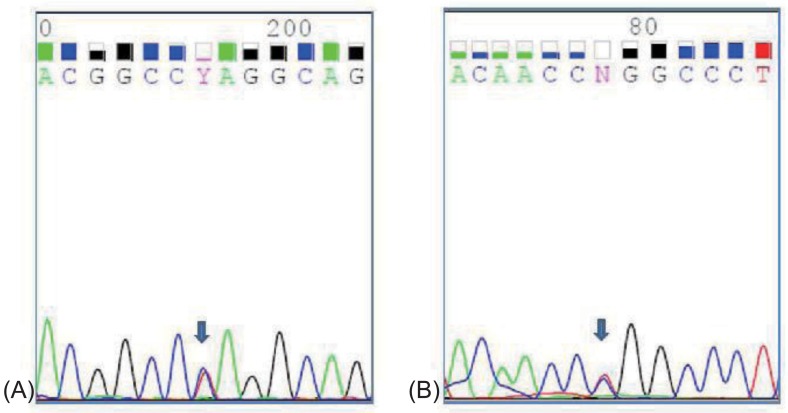
Heterozygous mutations were detected in exon 3 and 15 of SLC12A3 gene. (A) a heterozygous c.494A>T (p.Gln165Leu) in exon 3, (B) a heterozygous c.1868T>C (p.Leu623Pro) in exon 15.
The patient was treated with 0.7 IU/kg/wk of GH and administered oral replacement of magnesium oxide (MgO, 1 g) and potassium chloride (KCl, 1.8 g). Serum photassium and magnesium level increased to 3.0 mEq/L and 1.85 mg/dL. She showed a 5.6 cm height gain over seven months. The frequency of abdominal pain and diarrhea markedly decreased, and the tetany and muscle-cramping symptoms were eliminated.
Discussion
The patient, aged 12.9 years, presented with hypokalemia, metabolic alkalosis, and hyperreninemic hyperaldosteronism accompanied by short stature, obesity, and mild mental retardation. She was diagnosed with Gitelman syndrome by detection of heterozygous mutation in the SLC12A3 gene.
Gitelman syndrome results from various inactivation mutations in the SLC12A3 gene on chromosome 16q, which encodes the thiazide-sensitive sodium chloride cotransporter channel in the distal convoluted tubule. 172 distinct phenotype-variable mutations in the SLC12A3 gene have been reported4). Defects in the thiazide-sensitive NCCT impair Na+ and Cl- reabsorption, causing natriuresis and leading to volume contraction, which activates the renin-angiotensin-aldosterone axis. Increased aldosterone leads to secretion of K+ and H+, which causes hypokalemia and alkalosis5). Bartter syndrome also results from congenital renal tubule defects, the site of which is the thick ascending limb of the loop of Henle. Patients with Bartter syndrome present with symptoms in early childhood, and suffer a more severe failure to thrive. Patients with Gitelman syndrome, by contrast, are mostly asymptomatic or become fully symptomatic only in adolescence or adulthood6). Gitelman syndrome is associated with less severe growth retardation.
There are many factors that may contribute to growth retardation in Bartter or Gitelman syndrome. However, factors such as prematurity, significant salt loss in the antenatal-neonatal period, and marked polyuria are not present in Gitelman syndrome. Hypokalemia and metabolic alkalosis may have some effect on growth. Although hypokalemia can play key role in growth retardation, some patients still have growth problems after the normalization of serum electrolytes. Experimental studies, which found that rats fed a potassium-poor diet exhibited significant body-length reduction and weight gain, along with low levels of serum GH and insulin-like growth factor 1 (IGF-1), suggested that potassium depletion could have a negative effect on pituitary GH secretion7,8). Partial or complete GH deficiency has been found in several patients with either Bartter syndrome9-12) or Gitelman syndrome13-15). In all of the corresponding reports, growth response to GH treatment was generally good. In four reported cases of Gitelman syndrome with GH deficiency, growth velocity rose to 10.2-12 cm/yr during the first year of GH treatment. The girl in this report exhibited a 5.6 cm height gain during 7 months of GH treatment (growth velocity, 9.6 cm/yr). The reason for the association between hypokalemic tubulopathies and GH deficiency is not clear. GH treatment seemed to be helpful for the correction of hypomagnesemia. GH treatment has markedly increased magnesium absorption and retention in growing pigs16) and restored the serum magnesium levels in children with Gitelman syndrome13). In our case, as the patient was treated with MgO and GH simultaneously, the effect of GH on serum magnesium level could not be evaluated.
GH and IGF-1 treatment has been shown to not stimulate longitudinal growth unless to correct hypokalmeia in hypokalemic rats17,18). Experimental study demonstrated that potassium depletion leads to tissue-specific alteration in GH and IGF-1 metabolism in rats fed a potassium-deficient diet19).
Pubertal delay seems not to correlate with Gitelman syndrome symptoms. In this case, pubertal stage was delayed relative to chronologic age, but pubertal responses of LH and FSH to GnRH were observed.
If a child with Gitelman syndrome shows marked growth retardation, the GH-IGF-1 axis should be checked. GH treatment as well as the correction of serum potassium level is important for optimal growth. Further clinical research on Gitelman syndrome patients is required for more comprehensive analysis of the relations among growth retardation, abnormal GH-IGF-1 axis, and response to GH treatment.
Acknowledgments
This case report was supported by a grant (A120017) from the Korea Healthcare technology R&D Project, Ministry for Health, Welfare and Family Affairs, Republic of Korea.
Notes
No potential conflict of interest relevant to this article was reported.