Six-year follow-up of a child with familial chylomicronemia syndrome: disease course and effectiveness of gemfibrozil treatment --case report and literature review
Article information

Abstract
Familial chylomicronemia syndrome (FCS) is a rare autosomal recessive disease affecting lipoprotein metabolism. FCS is estimated to occur in 1 in 1–2 million individuals and can be diagnosed at any age, equally affecting all genders, races, and ethnicities. The condition is characterized by hypertriglyceridemia, which may predispose patients to acute pancreatitis. In this report, we present the case of a now 6-year-old girl with FCS on gemfibrozil and dietary restrictions. The patient initially presented at 40 days of age with vomiting. Serum samples revealed lipemia, with markedly elevated triglyceride levels. The patient was diagnosed with FCS, confirmed by genetic testing showing the homozygous variant c.833C>T(p,Ser278Phe) for the LPL gene. Despite being on a low-fat diet with medium chain triglyceride (MCT) based milk formulas, the patient developed acute pancreatitis 2 months later with continued elevated triglyceride levels. She was placed on gemfibrozil and fat-soluble vitamins at 2 months of age, with marked improvements subsequently noted. Currently, the patient is doing well, with normal growth parameters and no other episodes of acute pancreatitis. Her triglyceride levels have been maintained within normal levels. FCS is a rare, inherited lipid disorder that often goes underdiagnosed and unmanaged. It is worth considering the fibric acid derivative (gemfibrozil) to be one of the lines of management early on after diagnosis.
Highlights
· Familial chylomicronemia syndrome is a rare, inherited primary lipid disorder that is often underdiagnosed and mismanaged. There is insufficient data regarding its treatment protocol. The mainstays of management are dietary restrictions and lipid-lowering agents. This case report highlights the successful long-term use of gemfibrozil to treat a patient with this condition with promising early results.
Introduction
Familial chylomicronemia syndrome (FCS) is a rare metabolic disease characterized mainly by elevated triglyceride levels, which may predispose patients to recurrent pancreatitis, xanthomas, lipemia retinalis, and coronary heart disease. The condition is primarily caused by a defect in the lipoprotein lipase gene. However, other genetic mutations have also been identified. The treatment of FCS primarily includes dietary fat restrictions and the inclusion of MCTs. However, some severe cases have required lipid-lowering medications. Patients rarely require plasmapheresis, and new medications under trial have exhibited promising results. In this report, we also described the clinical features, genetic mutations, disease course, and management of FCS and provided a literature review of the condition.
Case report
This patient is a now 6-year-old girl who initially presented with vomiting at 40 days of age. Serum samples revealed lipemia, and the vital signs were normal upon physical examination. Weight, height, and head circumference were appropriate for her age, her abdomen was soft, and no dysmorphic features, skin abnormalities, or organomegaly were observed. The patient's initial lipid profile revealed normal total cholesterol of 4.2 mmol/L (normal total cholesterol < 4.4 mmol/L) and significantly high serum triglycerides at 36.4 mmol/L (normal TG [age 0–9 years] < 0.8 mmol/L) (Table 1).
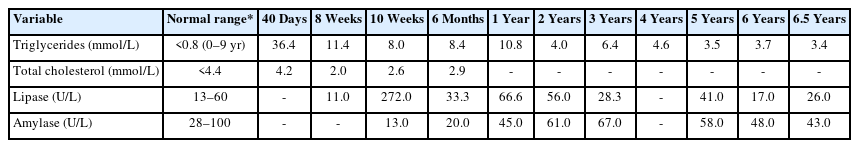
Laboratory results of the patient since diagnosis
A dietician was involved in her care, and she was placed on a low-fat diet with an MCT-based milk formula and cessation of breastfeeding. Upon follow-up, the patient's triglyceride and cholesterol levels had decreased, and she was clinically asymptomatic.
However, at the age of 2 months, she was readmitted with recurrent projectile vomiting. No fever, loose stools, or history of sick contact were noted, and her physical examination was unremarkable. In addition, the abdominal ultrasound was normal. The patient's lab results revealed low hemoglobin levels, high pancreatic enzymes, and high triglyceride levels (8.0 mmol/L). She was, therefore, diagnosed with acute pancreatitis due to hypertriglyceridemia and managed accordingly. The patient was managed as nil per os, intravenous (IV) fluids were initiated. She was placed on gemfibrozil 150 mg twice daily (70 mg/kg/day) and fat-soluble vitamin supplements. Her vomiting subsided, pancreatic enzyme levels normalized, and triglyceride levels decreased. The patient was discharged after 4 days with triglyceride levels of 4.4 mmol/L.
At 6 months of age, she was admitted with a fever and cough and was diagnosed with bronchopneumonia. Her lipid profile upon admission exhibited triglyceride levels of 15.87 mmol/L and pancreatic enzymes within normal ranges. The patient required oxygen support from a high-flow nasal cannula, IV antibiotics, and bronchodilator nebulization. Additionally, her hemoglobin levels were low on admission, requiring a transfusion. The enterovirus polymerase chain reaction was positive on the respiratory panel. The patient was discharged after 11 days with the gemfibrozil dose increased to 180 mg twice a day. Upon discharge, her triglyceride levels were 8.44 mmol/L.
The patient was born at term by spontaneous vaginal delivery with a birth weight of 2.8 kg. The antenatal history was unremarkable. Her parents are first-degree cousins, and she is the first child. Her mother has hypercholesterolemia managed with diet control, and both maternal and paternal grandmothers have hyperlipidemia and are on lipid-lowering medications. The patient's vaccination schedule is complete, and her development is normal for her age. She was seen by an ophthalmologist at 16 months of age and diagnosed with potential lipemia retinalis. The patient was advised for a follow-up and electroretinogram, performed at 6 years of age, revealing the same finding of lipemia retinalis. The patient is currently awaiting another follow-up after 1 year.
Molecular genetic analysis (DNA sequencing) of the LPL gene in exons 5 and 6 was positive for the homozygous variant c.833C>T(p,Ser278Phe) of unknown clinical significance. Both parents were heterozygous for the same variant. This variant is a potential disease-causing variant but has yet to be confirmed due to the inability to conduct a functional study.
The patient continued to be followed up in a pediatric endocrinology clinic. She remained asymptomatic, with fluctuating serial triglyceride levels, primarily within acceptable limits. The gemfibrozil dose was adjusted accordingly, reaching 600 mg twice a day. The patient also continued regular follow-ups with a dietitian who advised MCT oil-based formula (Monogen) and a low-fat diet. She was not compliant with the diet restrictions, as reflected by her fluctuating triglyceride levels.
Our reported patient is currently 6 years and 6 months old, asymptomatic, and has no history of other hospital admissions. Her weight is in the 8th percentile, and her height is in the 14th percentile, based on Center for Disease Control growth charts. She remains on a low-fat diet with MCT oil-based milk, Monogen, 400 mL/day. Her current medications include gemfibrozil 600 mg orally twice daily, vitamin D3 1,000 IU orally twice daily, omega 3–3.5 gm/day orally twice daily, and multivitamins. Her most recent triglyceride levels were noted at 3.4mmol/L (Fig. 1)
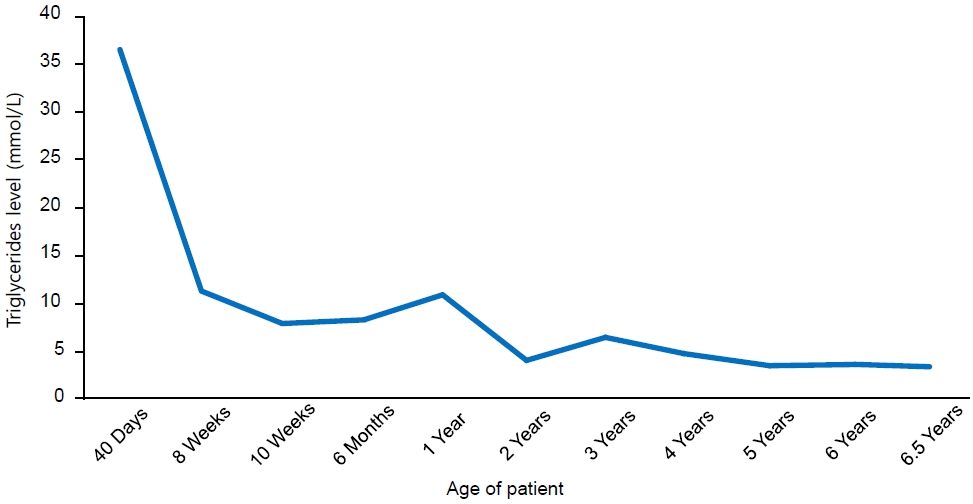
Trend of triglyceride levels of the patient since diagnosis.
Our patient's close clinical follow-ups and laboratory monitoring revealed no side effects from gemfibrozil, and the risk of acute pancreatitis was significantly reduced.
Informed and written consent was obtained from the parents to publish the details of their child's medical case and any accompanying images.
Discussion
We present a case of an early presentation of FCS, a rare genetic mutation, which did not respond to traditional treatment. The patient was initiated early in life to gemfibrozil and responded well. Dyslipidemias are disorders of lipoprotein metabolism. The overall prevalence of high non–high-density lipoprotein cholesterol is 10.7% in children aged 6–19 years [1]. Dyslipidemia is classified into primary and secondary types, with primary dyslipidemia being associated with either the overproduction and/or impaired removal of lipoproteins [2].
FCS is a rare primary dyslipidemia, with a prevalence of approximately 1 in 1–2 million [3]. FCS can be diagnosed at any age and can affect any sex, race, or ethnicity [4]. FCS is characterized by lipoprotein metabolism dysfunction due to a defect in lipoprotein lipase, an apolipoprotein C deficiency, or the presence of inhibitors to lipoprotein lipase [5]. Lipoprotein lipase enzymes cannot degrade triglycerides in the chylomicrons, leading to elevated triglyceride levels in the blood, even when fasting. Hypertriglyceridemia is defined as triglyceride levels above the 95th percentile of the corresponding age and sex [5]. The condition can manifest as eruptive xanthoma, lipemia retinalis, hepatosplenomegaly, and acute pancreatitis, the most severe manifestation, associated with 5%–6% overall mortality [5].
Symptoms are primarily present in childhood. However, 25% of cases may present in infancy, before one year of age [6]. In our case, the patient presented with vomiting and lipemic serum samples at 40 days old. In the literature, we found case reports describing patients with heterozygous mutations in the LPL gene presenting with pallor, jaundice, diarrhea, and irritability in early infancy between the ages of 20–60 days old [2,7]. Additionally, a 2021 study in India found that among 15 affected infants with severe hypertrigleceridemia (TG >12 mmol/L), 2 were symptomatic, with 1 patient presenting with chylothorax and the other presenting with acute pancreatitis. The other patients in that study had incidental findings of lipemia [8]. Another interesting manifestation of FCS was reported in a 24-day-old neonate who presented with hematuria [2]. Moreover, in 2 Korean cases reported, recurrent respiratory infections were observed in patients with elevated blood lipid levels at 11 and 28 days old [9].
Eighty percent of FCS cases are caused by biallelic mutations of the LPL gene located in chromosome 8p22. The remaining mutations are attributed to the ApoA5, GPIHBP1, ApoC2, and LMF1 genes [2]. Our patient's molecular genetic analyses of the LPL gene revealed the homozygous variant c.833C>T (p.Ser278Phe) of unknown clinical significance. This variant was listed in the Human Gene Mutation Database as a potential disease-causing variant since a pathogenic variant reported in the same codon position (p.Ser278Phe) exists [10]. Therefore, although this variant appears to be rare, it may cause the rare FCS disorder when the clinical and laboratory features of the patient are considered. In a retrospective study performed in India where 15 patients were evaluated with incidental detections of serum lipemia, genetic screening analysis of the LPL gene was offered to 14 patients (1 patient was lost to follow-up). In this study, 6 different variants were identified, of which 2 were novel variants [8]. In 2017, the Journal of Atherosclerosis Supplements presented a diagnostic algorithm for FCS to test LPL activity and assess the pathogenicity of undescribed variants, as novel LPL mutations are continuously being discovered [11].
In Saudi Arabia, 2 siblings were reported to have FCS, with the first sibling presenting at 2 days old with incidental lipemia while being investigated for neonatal jaundice. Triglyceride levels of 80 mmol/L were noted. The second sibling was then screened for FCS at 16 days old due to positive family history, and triglyceride levels of 5.92 mmol/L were noted [12].
The mainstay treatment of FCS is dietary restrictions of fat intake of no more than 20–25 g/day [12] and the addition of MCT, as they are water-soluble and absorbed directly by the portal vein without being incorporated into chylomicrons. In infants, breastfeeding should be discontinued and substituted with MCT-based formula.
Fibric acid derivatives, such as gemfibrozil, are effective medications to treat hypertriglyceridemia in adults. These derivatives lower triglyceride levels by modulating the activity of peroxisome proliferator-activated receptor-α in the liver with reduced hepatic secretion of very-low-density lipoproteins and increased lipolysis of plasma triglycerides [13]. Currently, no standard guidelines exist for using gemfibrozil in FCS. However, several reports exist on its use in pediatric patients with FCS. For instance, in Saudi Arabia, a 2-day-old male infant diagnosed with FCS was placed on gemfibrozil and pravastatin at the age of 60 days in addition to dietary restrictions [12]. This patient was admitted twice, at the age of 6 months and 2 years, where he presented with vomiting and loose stool due to acute gastroenteritis. The patient was managed accordingly, had no further attacks of abdominal pain requiring admissions, and was regularly followed up. His sibling was also diagnosed at the age of 16 days and was initially managed by dietary restrictions alone. Upon follow-up, his triglyceride levels were significantly elevated at 10.5 mmol/L. Therefore, gemfibrozil was initiated in this sibling at the age of 6 months [12]. In another case, a 2-month-old boy presented with irritability, abdominal distension, hepatosplenomegaly, and anemia. His initial serum sample was milky and viscous, and his triglyceride levels were substantially elevated at 150.07 mmol/L, serum cholesterol was at 56.89 mmol/L, and he was subsequently diagnosed with FCS. After dietary modifications, lipid lowering agents, and iron supplements, the patient's repeat serum sample after 15 days was red in color and his lipid profile had improved considerably with triglyceride levels of 88.3 mmol/L and total cholesterol of 25.4 mmol/L [7]. Moreover, in 2018, a case was reported on a 23-day-old boy who had an incidental lipemic sample exhibiting triglyceride levels of 116.39 mmol/L. Genetic testing confirmed FCS due to LPL mutation with homozygous missense variation at nucleotide position 478 (c. 478C>T) observed on exon 4. The patient was managed dietarily and placed on gemfibrozil. At the age of 2 months, his triglyceride levels had decreased to 9.85 mmol/L [14].
In conclusion, FCS is a rare, inherited primary lipid disorder that often goes underdiagnosed and unmanaged. The condition mainly presents in childhood but can also be observed in infancy with various manifestations. There is insufficient data regarding its treatment protocol. However, the mainstays of management are dietary restrictions and lipid-lowering agents. Notably, a fibric acid derivative agent, gemfibrozil, has been used to treat those patients with promising results early.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: MM; Data curation: MM; Formal analysis: MM; Methodology: MM; Project administration: MM; Visualization: MM; Writing - original draft: MM, MA; Writing - review & editing: MM