Introduction
Short stature in children is defined as a height of at least 2 standard deviations less than the mean for children of the same chronological age and sex. Longitudinal bone growth is primarily mediated by the growth plate, a specialized cartilaginous structure. Endochondral ossification is a complex phenomenon that promotes bone elongation and increasing height and involves proliferation, chondrocyte senescence and hypertrophy, and cartilage matrix synthesis [1]. Paracrine and autocrine factors are the primary regulators of endochondral ossification, and mutations in these genes can cause short stature [2].
Aggrecan, which is encoded by ACAN, is a primary proteoglycan component of the extracellular matrix of the growth plate and articular cartilage and contributes to the growth of plate cartilage. ACAN mutations were initially reported as 2 rare types of skeletal dysplasia: autosomal-dominant spondyloepiphyseal dysplasia, Kimberley type (SEDK, OMIM 608361) in 1990 [3], and autosomal-recessive spondyloepimetaphyseal dysplasia, aggrecan type (SEMD, OMIM 612813), which was previously reported in only one family [4]. A heterozygous ACAN mutation has been reported as a major cause of idiopathic short stature and familial short stature. However, only one case of a young child without early advanced bone age, which is a typical manifestation of ACAN mutation, has been reported in Korea [5]. Here, we describe the case of an adolescent boy with severely short stature who harbored a heterozygous c.512C>T (p.Ala171Val) mutation of ACAN, which has not yet been reported to be associated with clinical manifestation.
Case report
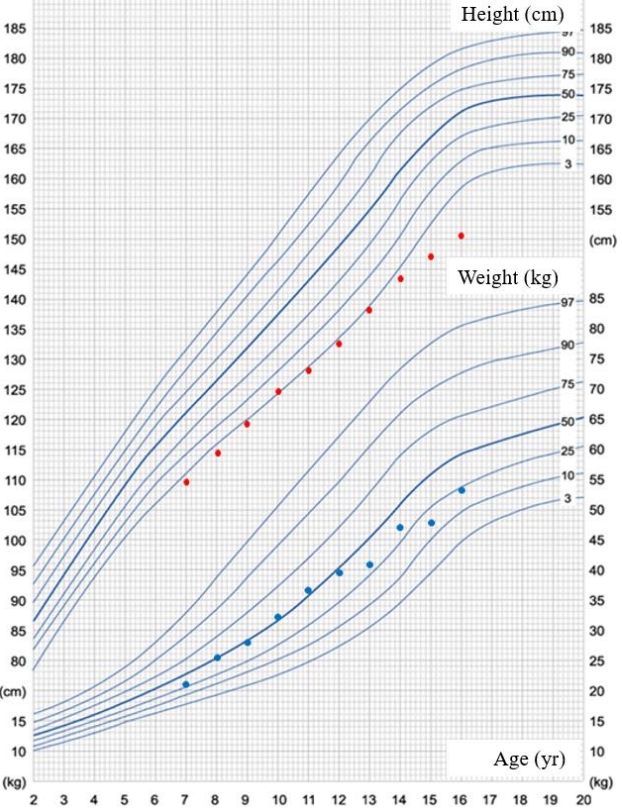
A 15-year-old boy presented to Yeungnam University Hospital with severely short stature. He was the first of 3 children born to nonconsanguineous parents. His height and weight were 149 cm (Korean standard deviation score [SDS] of -3.6) and 50.5 kg (-1.48 SDS), respectively. He was born at 39 weeks through normal spontaneous vaginal delivery and with a birth weight of 3.2 kg. When we reviewed his past school physical examination records, we noted short stature and growth retardation during childhood (Fig. 1). He also presented with mild midfacial hypoplasia, frontal bossing, a broad chest, a short neck, ptosis, brachydactyly, and a low posterior hairline (Fig. 2A, B). Physical examination revealed a Tanner stage 4 and a 15- to 20-mL bilateral testicular volume. Laboratory serum analysis provided the following findings: hemoglobin, 14.6 g/dL; leukocytes, 9,070/┬ĄL; platelet count, 341├Ś103/mm; aspartate aminotransferase, 31 IU/L; alanine aminotransferase, 19 IU/L; sodium, 138 mEq/L; potassium, 4.0 mEq/L; blood urea nitrogen, 11.1 mg/dL; and creatinine, 0.92 mg/dL. Serum levels of insulin-like growth factor 1 and insulin-like growth factorbinding protein-3 were 486.3 ng/mL (normal range, 133ŌĆō828 ng/mL) and 6,110 ┬Ąg/mL (normal range, 3,287ŌĆō7,735 ┬Ąg/mL), respectively, and the T3, free T4, and TSH levels were 1.68 nmol/L (normal range, 1.2ŌĆō2.8 nmol/L), 12.63 pmol/L (normal range, 10ŌĆō20 pmol/L), and 1.34 mu/L (normal range, 0.3ŌĆō4 mu/L). No endocrine abnormalities were detected. His bone age was 17ŌĆō18 years and was determined using the standard Greulich-Pyle method, which was advanced compared with his chronological age of 15 years 3 months (Fig. 2C). Pelvic and spinal x-ray imaging did not reveal skeletal dysplasia.
His father and grandfather also had short stature. His father's and mother's heights were 150 cm (-4.8 SDS) and 153 cm (-1.69 SDS), respectively. His younger brother and sister were 13 and 12 years old and had heights of 167 cm (0.36 SDS) and 157 cm (0.90 SDS), respectively. They did not present any other abnormal clinical manifestations. The patient's father exhibited ptosis and a short neck (Fig. 2D) and had undergone pulmonary valvuloplasty at our hospital and had received treatment for lumbar disc herniation in his 30s. Therefore, next-generation sequencing was carried out to detect Noonan syndrome; however, negative findings were obtained.
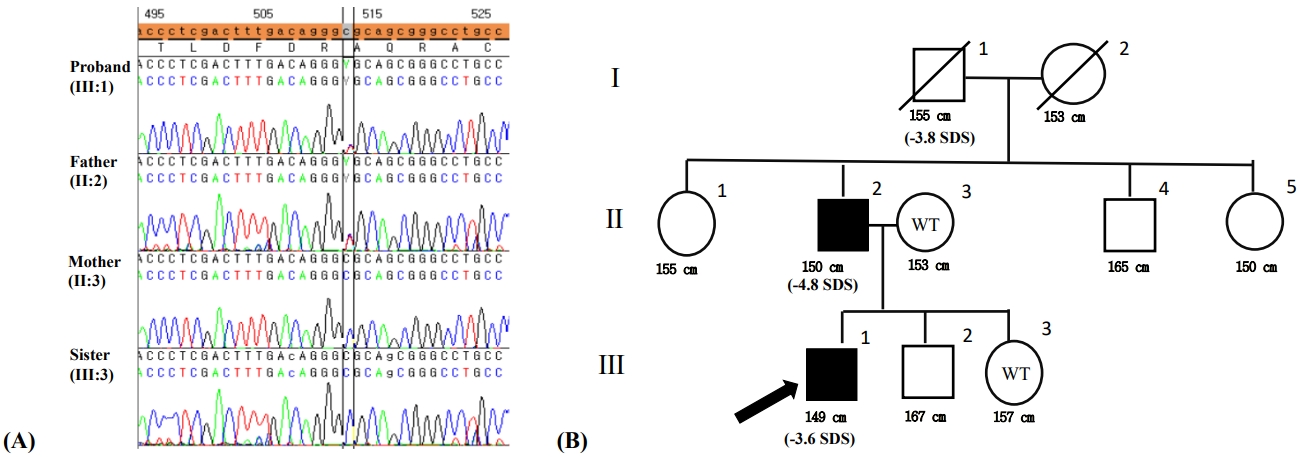
After obtaining written informed consent for genetic testing, whole-exome sequencing was performed. Genomic DNA was extracted from saliva and buccal swab samples from the patient. All exon regions of all genes (~22,000) were detected using the SureSelect kit (Agilent, SantaClara, CA, USA) in accordance with the manufacturer's instructions and were sequenced with the Illumina platform (NovaSeq, Illumina, San Diego, CA, USA). Data analysis for raw genome sequences (including alignment with the GRCh37/hg19 human reference genome, variant calling, and annotation) were conducted using in-house software. Variants with minor allele frequencies less than 0.05% for dominant disease associations or 2% for recessive diseases in population genome databases, including the Exome Variant Server (http://evs.gs.washington.edu/EVS/), the 1000 Genomes (http://phase3browser.1000genomes.org), and ExAC (http://exac.broadinstitute.org/), were considered for further interpretation. The proband's clinical profile was accessed to assess similarity with ~7,000 genetic diseases. The pathogenicity of each variant and its associated diseases was evaluated in accordance with the ACMG/AMP guidelines without adjustment of criteria weighting. Heterozygous variant c.512C>T was detected in exon 4 in ACAN (p.Ala171Val) (Fig. 3A). This variant was considered potentially pathogenic in accordance with the PM1, PM2, PP3, and PP5 criteria. This variant is located in the link domain of ACAN, which is a well-established functional domain (PM1) that has been reported at an extremely low frequency in the aforementioned population databases (PM2); is predicted to have a deleterious effect on the gene product via multiple in silico prediction tools (DANN [score 0.9989], EIGEN [0.9592], PrimateAI [0.7016], REVEL [0.509], and MetaSVM [0.1913]) (PP3); and is registered as "likely pathogenic" in the ClinVar database (PP5).
Direct sequencing was performed for the patient, his parents, and his younger sister, and the regions of interest were determined. His mother and younger sister displayed the wild type sequence, while the heterozygous mutation was identified in his father (Fig. 3B).
Discussion
ACAN is located on chromosome 15q26.1 and comprises 1ŌĆō18 exons and the untranslated regions [6,7]. Aggrecan, which is encoded by ACAN, contains a 250-kDa protein core with approximately 100 chondroitin sulfate and 30 keratan sulfate glycosaminoglycan chains. It is linked to 3 globular domains through one large domain. These globular domains regulate interactions with other components of the extracellular matrix [8]. Aggrecanopathies have emerged as a phenotype of genetic skeletal disease in humans. Aggrecan-related bone diseases include 5 clinical phenotypes: spondyloepimetaphyseal dysplasia, aggrecan type (OMIM 612813); macrocephaly with multiple epiphyseal dysplasia (OMIM 607131); spondyloepiphyseal dysplasia, Kimberley type (OMIM 608361); familial osteochondritis dissecans, short stature, and early-onset osteoarthritis (OMIM 165800); and various idiopathic short stature phenotypes. Autosomal-dominant short stature, premature growth cessation, and accelerated bone age maturation were reported in 3 families in 2014 [9]; one family displayed a heterozygous missense mutation (c.7064 T>C; p.Leu2355Pro), while the second family had a base-pair substitution (c.2026+1G>A) at the splice donor site on exon 10, resulting in skip of exon 10. The third family displayed a frameshift mutation (c.272delA). Since then, more than 100 individuals with autosomal-dominant inherited short stature have been reported as carriers of ACAN mutations [10]. The phenotypic spectrum of heterozygous ACAN mutations ranges from mild and proportionate short stature to mild skeletal dysplasia with disproportionate short stature with no genotype-phenotype correlations. ACAN mutations potentially reflect mild dysmorphological findings similar to the present case, including midfacial hypoplasia, relative macrocephaly, a flat nasal bridge, frontal bossing, broad thumbs, lordosis, and brachydactyly [10]. Phenotypes may differ widely even within the same family. This phenotype spectrum suggests a dose effect with a range of penetrance. Functional aggrecan haploinsufficiency results in premature hypertrophic chondrocyte maturation and early invasion of blood vessels and osteoblasts in the growth plate, which have been proposed as a mechanism underlying advancement of bone age, premature growth cessation, and early epiphyseal fusion in patients harboring ACAN mutations [11,12]. Similar to the present case, early disc herniation is reportedly associated with ACAN mutations [10,13]. A cartilage matrix-deficient mice model induced by absence of aggrecan revealed that ACAN mutations result in early-onset and multiple spinal disc herniations owing to a reduction in aggrecan level in the cartilage and accelerated degeneration of disc cartilage [14]. Affected patients have advanced bone age at prepubertal stages and present with premature growth cessation after puberty [9,10,13]. Van der Steen et al. [15] reported that ACAN mutations apparently influence growth impairment even before birth; these mutations were identified in a cohort study of children born small for gestational age. Gkourogianni et al. [10] reported that the median-height SDS of individuals with ACAN mutations was -2.8 in adulthood and -2.0 in childhood; and the mean difference in advanced bone age (bone ageŌĆōchronologic age) was +1.3 years. Although these patients are not associated with precocious puberty, suppression of puberty with a gonadotropin releasing hormone (GnRH) analog might be beneficial to block early growth cessation. Recent reports have indicated the effectiveness of a combined treatment that includes a GnRH analog and growth hormone (GH) to help patients who harbor ACAN mutations achieve an appropriate adult height [9,10,15]. A previous study reported a height increase of 5ŌĆō8 cm among patients receiving combinatorial GnRH and GH treatment in comparison with sex-matched family members [15].
In summary, short stature is generally associated with delayed bone age involving GH deficiency, hypothyroidism, Cushing syndrome, and idiopathic short stature. Short stature combined with advanced bone age is much less common. Moreover, recent large-scale cohort studies of ACAN mutations have been conducted worldwide with targeted exome sequencing, and new ACAN mutations have been identified [16-18]. This case suggests that ACAN mutations are the most likely etiology among patients with idiopathic short stature and advanced bone age and thus warrant early treatment.