The 46,XX testicular disorder of sex development (DSD), also known as 46,XX male syndrome, is a rare form of DSD and clinical phenotype shows complete sex reversal from female to male. The sex-determining region Y (SRY) gene can be identified in most 46,XX testicular DSD patients; however, approximately 20% of patients with 46,XX testicular DSD are SRY-negative. The SRY-box 9 (SOX9) gene has several important functions during testis development and differentiation in males, and overexpression of SOX9 leads to the male development of 46,XX gonads in the absence of SRY. In addition, SOX9 duplication has been found to be a rare cause of 46,XX testicular DSD in humans. Here, we report a 4.2-year-old SRY-negative 46,XX boy with complete sex reversal caused by SOX9 duplication for the first time in Korea. He showed normal external and internal male genitalia except for small testes. Fluorescence in situ hybridization and polymerase chain reaction (PCR) analyses failed to detect the presence of SRY, and SOX9 intragenic mutation was not identified by direct sequencing analysis. Therefore, we performed real-time PCR analyses with specific primer pairs, and duplication of the SOX9 gene was revealed. Although SRY-negative 46,XX testicular DSD is a rare condition, an effort to make an accurate diagnosis is important for the provision of proper genetic counseling and for guiding patients in their long-term management.
XX male syndromeSex determining region Y-box 9Sex-determining region gene on YDisorders of sex development
Introduction
The 46,XX testicular disorder of sex development (DSD), also known as 46,XX male syndrome, is a rare condition with an estimated prevalence of one in 20,000 males1). It can be classified into two subgroups, SRY-positive or SRY-negative, according to the presence or absence of the sex-determining region Y (SRY) gene. Approximately 80% of patients with 46,XX testicular DSD have SRY on one of two X chromosomes, which results from abnormal chromosomal translocation during gametogenesis. However, the remaining 20% of individuals with 46,XX testicular DSD are SRY-negative, and the cause of this condition is largely unknown1). Most individuals with 46,XX testicular DSD with SRY have normal genitalia and are usually not diagnosed until puberty does not proceed normally. However, SRY-negative individuals are more likely to have ambiguous genitalia than SRY-positive individuals. Therefore, 46,XX SRY-negative individuals showing complete male genitalia are quite rare2).
The development of testes from bipotential gonadal primordia is controlled by complex molecular networks of expressions of multiple genes. Among the genes, SRY on the Y chromosome is a typical male-specific gene, which initiates testis development and directly upregulates the SRY-related HMG box-containing gene 9 (SOX9) gene expression. SOX9 is a transcription factor with a DNA-binding site very similar to SRY, and plays a crucial role in the cascade of gene interactions for differentiation and development of the testis3). In 1999, SOX9 overexpression in a 46,XX gonad was identified as leading to an induction of male genitalia in the absence of SRY4). Although there have been a few reports to date of 46,XX SRY-negative patients having duplication of SOX94,5,6,7), SOX9 duplication is not a common cause of 46,XX testicular DSD8). In Korea, less than 10 SRY-positive 46,XX patients have been reported, and there are no reports of 46,XX testicular DSD resulting from SOX9 duplication9).
In this study, we present an SRY-negative 46,XX Korean boy who presented with small testes and without any genital ambiguity. Molecular genetic studies confirmed SOX9 duplication as the cause of 46,XX testicular DSD for the first time in Korea.
Case report
1. Subject
A 4.2-year-old boy was referred to the outpatient clinic at Seoul National University Children's Hospital for evaluation of an abnormal karyotype, 46,XX. Four months before the visit, his mother noticed that his testes were smaller than those of his newborn brother. He visited the Pediatric Urology Clinic at a regional hospital, and karyotype analysis using his peripheral blood resulted in a diagnosis of 46,XX. He was born at a gestational age of 40 weeks by vaginal delivery, with a birth weight of 3.2 kg (25th-50th percentile), without any perinatal problems. After birth, a small atrial septal defect was detected, which was closed spontaneously on follow-up.
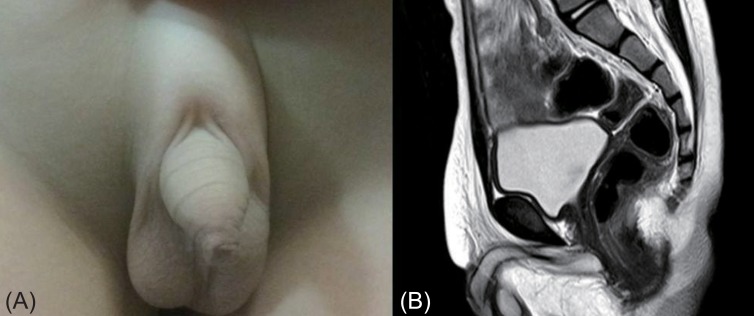
On physical examination, his body weight and height were 14.9 kg (10th-25th percentile) and 98.2 cm (10th-25th percentile), respectively. His head circumference was 47.5 cm (5th percentile), and he showed normal developmental milestones. No gross anomalies were observed, and his external genitalia showed complete male phenotype without ambiguity (Fig. 1A). The phallus was 3.0 cm long, and the opening of the urethral meatus was located at the tip of the glans penis. The perineal surface between the urethral meatus and the anal opening was closed. The scrotum was bifid with small palpable gonads. The longest testicle was less than 1 cm. No skeletal abnormalities were identified. His bone age was 4 years, and magnetic resonance imaging revealed that both testes were located in the scrotal sac; no structure like the uterus, ovary, or vagina was found (Fig. 1B).
Baseline serum concentrations of luteinizing hormone and follicle-stimulating hormone were <0.5 mIU/mL (prepubertal normal range, 0.02-1.03 mIU/mL) and 0.9 mIU/mL (prepubertal normal range, 0.25-1.92 mIU/mL), respectively. The estradiol level was 4 pg/mL (normal range for boys, 3-10 pg/mL), and the anti-Müllerian hormone level was 44.0 ng/mL (normal range for boys, 7-240 ng/mL). Levels of 17-hydroxyl progesterone and dehydroepiandrosterone sulfate were within normal ranges. The results of a human chorionic gonadotropin (hCG) stimulation test to assess testicular function showed a normal response: the baseline testosterone level was <0.1 ng/dL (normal range, <0.12 ng/dL), and the follow-up testosterone level after intramuscular injections for 3 consecutive days with 1,000 IU of hCG elevated to 2.10 ng/dL.
2. Genetic analyses
The parents provided formal informed consent to permit peripheral blood sampling for genetic testing, and the Institutional Review Board of Seoul National University Hospital approved this study. We repeated karyotype analysis using the patient's blood lymphocytes. Twenty cells were analyzed, and the karyotype was identified as 46,XX10).
Fluorescence in situ hybridization (FISH) analysis to identify the presence of the SRY gene on an X chromosome was conducted; SRY was not revealed. Polymerase chain reaction (PCR) and electrophoresis were performed using specific primers for SRY; however, we could not find any band of amplified SRY materials.
We performed additional molecular genetic analyses for assessing the SOX9 gene. Direct sequencing analysis for all coding exons and exon-intron boundaries of SOX9 did not identify any pathogenic mutations. Therefore, a real-time PCR assay was subsequently performed to identify the copy number of SOX9, using two sets of primer pairs. Each primer pair was designed to be located in the region approximately 5,000 bp upstream and downstream from the SRY gene. We used genomic DNA from a normal fertile 46,XY male as a control, and real-time PCR analyses with each primer pair were repeated three times. The results of real-time PCR reactions showed a duplicated copy number of SOX9 in the patient: the ratio of the patient to the control male was 1.45:1 (using an upstream primer pair) and 1.44:1 (using a downstream primer pair) (Fig. 2).
Discussion
46,XX male syndrome was first described in 196411), and the Chicago consensus renamed of this syndrome as 46,XX testicular DSD in 200512). Most patients have normal male phenotypes at birth, and are usually diagnosed in late adolescence because of delayed puberty, gynecomastia, or infertility2). Approximately 15% of XX males have hypospadias, cryptorchidism, or more severe genital ambiguity13). All 46,XX males are infertile owing to the absence of the azoospermia factor region in the long arm of the Y chromosome14,15). Although testosterone levels normally evolve during puberty, deficient testosterone production after a β-hCG stimulation test becomes evident and hypergonadotropic hypogonadism with severe testicular atrophy and azoospermia in biopsied testes is clearly manifested in adulthood15,16). In this case, there was normal response in the plasma testosterone level after β-HCG stimulation performed at the age of 4.2 years, and this result only suggested that testis tissue was present. As testicular atrophy progresses with age, follow-up stimulation test performed during adolescence might show abnormal response as testicular atrophy progresses. Testosterone replacement therapy may be required at puberty.
Female-to-male sex reversal in individuals with 46,XX is most frequently caused by the presence of SRY on one of the X chromosome resulting from a translocation between the X and Y chromosomes17). This translocation event occurs randomly during spermatogenesis in the affected individual's father. The SRY gene plays a critical role in the initiation of testicular differentiation in males18). In XY embryos, SRY, which is normally located on the Y chromosome, induces the gonadal primordia to develop into testes. If a fetus is conceived from a sperm with an SRY-bearing X chromosome, it will develop as a male despite not having a Y chromosome.
In about 80% of individuals with 46,XX testicular DSD, the presence of SRY can be documented by FISH or PCR analyses and classified as SRY-positive; the remaining 20% without SRY are classified as SRY-negative. Some genotype-phenotype correlations have been suggested, and SRY-negative individuals have a higher incidence of genital ambiguity, which is not common in SRY-positive individuals2,17).
Because testicular differentiation occurs without SRY in approximately 20% of individuals with 46,XX testicular DSD, there might be other important genes that cause 46,XX testicular DSD19). SOX9 on chromosome 17q24.3 is one of the genes that plays an important role in the development of the skeleton and genital organs4). During embryogenesis, SOX9 is expressed immediately downstream of SRY and functions as a critical Sertoli cell differentiation factor. In addition, differentiated Sertoli cells express anti-Müllerian hormone, which is required for Müllerian duct regression and differentiation of male genitalia14).
A family study recently reported that 46,XY persons with a 178 kb duplication in a region about 500 kb upstream of SOX9 were completely normal fertile males, whereas 46,XX SRY-negative persons in the same family with duplications were clinically infertile males6). In this report, two of the three paternal cousins of their probands are known as infertile and the two probands share the same paternal haplotype for the SOX9 region, confirming the possibility that the father was indeed the carrier of the same triplication. Therefore, we should investigate family members of 46,XX male patients caused by SOX9 duplication.
Male sex differentiation is known to require a delicate dosage balance among male determining genes, including SOX9. Haploinsufficiency of SOX9 from the loss of function mutations or deletions results in campomelic dysplasia (Online Mendelian Inheritance in Man #114290), which is a well-known skeletal dysplasia showing congenital bowing of long bones with male-to-female sex reversal in two-thirds of affected individuals with 46,XY20). On the other hand, duplication of SOX9 has been found in individuals with SRY-negative 46,XX testicular DSD. Previous studies have suggested that overexpression of SOX9 could induce testis determination in the absence of SRY4). In the present study, we also revealed SOX9 duplication as the cause of SRY-negative 46,XX testicular DSD by real-time PCR in a boy who presented with small testes and without any genital ambiguity. To our knowledge, this is the first report in Korea of 46,XX testicular DSD caused by SOX9 duplication.
However, the results of a recent study suggested that duplication of SOX9 is not a common cause of 46,XX testicular or 46,XX ovotesticular DSD8). In that study, the authors amplified microsatellite markers in the region of SOX9 from a cohort of 30 patients with either 46,XX testicular or 46,XX ovotesticular DSD to detect SOX9 duplications, but they could not find any duplication of the SOX9 region in 17q. They implied that there might be another important cause of 46,XX testicular DSD8). Another gene, SOX3, has recently been identified as upregulating the expression of SOX9 via a similar mechanism to SRY and as being responsible for XX male sex reversal in humans through gain-of-function mutations mediated by genomic rearrangements around SOX3, possibly leading to its altered regulation10). Furthermore, duplications of the SOX3 locus were detected in three cases of 46,XX testicular DSD10). SOX3 analysis can be considered the next step for the diagnosis of 46,XX SRY-negative testicular DSD with normal copies of SOX9.
Human sex determination and differentiation are complex processes associated with multiple genes including SRY, SOX9, SOX3, DAX-1, SF-1, and WT-14). However, the exact mechanism underlying male sex determination is not completely understood yet, and multiple genes associated with testicular differentiation are not routinely or fully investigated in most cases of 46,XX testicular DSD. Although SRY-negative 46,XX testicular DSD is a rare condition, an effort to make an accurate diagnosis is important for the provision of proper genetic counseling and for guiding patients in their long-term management.
No potential conflict of interest relevant to this article was reported.
BoucekkineCToublancJEAbbasNChaabouniSOuahidSSemrouniMClinical and anatomical spectrum in XX sex reversed patients: relationship to the presence of Y specific DNA-sequencesClin Endocrinol (Oxf)1994407337428033363ZentenoJCLopezMVeraCMendezJPKofman-AlfaroSTwo SRY-negative XX male brothers without genital ambiguityHum Genet19971006066109341880OnoMHarleyVRDisorders of sex development: new genes, new conceptsNat Rev Endocrinol20139799123296159HuangBWangSNingYLambANBartleyJAutosomal XX sex reversal caused by duplication of SOX9Am J Med Genet19998734935310588843CoxJJWillattLHomfrayTWoodsCGA SOX9 duplication and familial 46,XX developmental testicular disorderN Engl J Med2011364919321208124VetroACicconeRGiordaRPatricelliMGDella MinaEForlinoAXX males SRY negative: a confirmed cause of infertilityJ Med Genet20114871071221653197BenkoSGordonCTMalletDSreenivasanRThauvin-RobinetCBrendehaugADisruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex developmentJ Med Genet20114882583022051515SeeherunvongTUkarapongSMcElreaveyKBerkovitzGDPereraEMDuplication of SOX9 is not a common cause of 46,XX testicular or 46,XX ovotesticular DSDJ Pediatr Endocrinol Metab20122512112322570960LeeJWHongCHKimHRSinJBA case of SRY negative 46, XX male syndrome with deletion on long arm of X chromosomeKorean J Perinatol200617353358MoalemSBabul-HirjiRStavropolousDJWherrettDBagliDJThomasPXX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplicationAm J Med Genet A2012158A1759176422678921De la chapelleAHortlingHNiemiMWennstroemJXX chromosomes in a human male. First caseActa Med Scand1964175Suppl 412252814154995LeePAHoukCPAhmedSFHughesIAInternational Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric EndocrinologyConsensus statement on management of intersex disorders. International Consensus Conference on IntersexPediatrics2006118e488e50016882788de la ChapelleAAnalytic review: nature and origin of males with XX sex chromosomesAm J Hum Genet197224711054622299De Santa BarbaraPBonneaudNBoizetBDesclozeauxMMoniotBSudbeckPDirect interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Mullerian hormone geneMol Cell Biol199818665366659774680Ergun-LongmireBVinciGAlonsoLMatthewSTansilSLin-SuKClinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literatureJ Pediatr Endocrinol Metab20051873974816200839Maciel-GuerraATde MelloMPCoeliFBRibeiroMLMirandaMLMarques-de-FariaAPXX Maleness and XX true hermaphroditism in SRY-negative monozygotic twins: additional evidence for a common originJ Clin Endocrinol Metab20089333934318056774de la ChapelleAHastbackaJKorhonenTMaenpaaJThe etiology of XX sex reversalReprod Nutr Dev1990Suppl 139s49s1976312SinclairAHBertaPPalmerMSHawkinsJRGriffithsBLSmithMJA gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motifNature19903462402441695712McElreaveyKVilainEAbbasNHerskowitzIFellousMA regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male developmentProc Natl Acad Sci U S A199390336833728475082WagnerTWirthJMeyerJZabelBHeldMZimmerJAutosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9Cell199479111111208001137
(A) The genitalia photograph of the patient was taken at 4.2 years of age. His external genitalia show complete male phenotypes without ambiguity. (B) Normal internal genital organs are visible, and there is no evidence of female genital organs on the T2-weighted magnetic resonance image.
Results of real-time polymerase chain reaction analysis using two sets of specific primer pairs (A, proximal primer pair; B, distal primer pair) show the increased copy number of SOX9 in the patient (approximately 1.5 times) compared with a control male. RQ, relative quantification.