Highlights
┬Ę We report a male aged 4 years 7 months with obese Wilson's disease (WD) accompanied by nonalcoholic fatty liver disease. We diagnosed him with WD incidentally based on targeted gene panel sequencing to screen for genes related to pathologic obesity. Early suspicion and diagnosis are important in this treatable and reversible disease.
Introduction
Wilson disease (WD, OMIM #277900), or progressive hepatolenticular degeneration, is an autosomal recessive inherited disorder affecting copper metabolism whose incidence is estimated to be 1/30,000 in most populations [1]. In this disease, copper secretion from the liver into the bile fails, and copper is incorporated into ceruloplasmin. Copper primarily accumulates in the liver and brain tissue, so this disease usually manifests early as chronic liver disease and later as a neurological disorder. Hepatic manifestations can include any type of liver disease, from acute to chronic. WD is fatal if left untreated, but timely treatment can prevent severe liver disease and lifelong neurologic disabilities.
WD is usually suspected if the serum ceruloplasmin is low in patients with elevated liver enzyme levels, and its diagnosis relies on detection of Kayser-Fleischer rings, low ceruloplasmin, elevated urine and hepatic copper levels, signs of liver or neurologic disease, or associated histologic liver changes. Recent studies have reported on diagnosed WD cases with atypical symptoms through genetic testing to identify homozygous or complex heterozygous mutations in the ATP7B gene.
Obesity is not a common feature in WD, is induced by decrease in energy expenditure, and has variable phenotypic severity. Common characteristics of obesity include insulin-resistance, hyperlipidemia, and nonalcoholic fatty liver disease (NAFLD). NAFLD is a spectrum of liver diseases strongly associated with obesity and is the most common chronic liver disease in children. It is also commonly observed in WD, and it is the most frequently observed source of hepatic lesions [2]. Copper accumulation might be related to NAFLD pathogenesis [3].
Here we report a boy diagnosed with WD via genetic testing for severe obesity, along with a comprehensive literature review of the endocrine complications associated with WD.
Case report
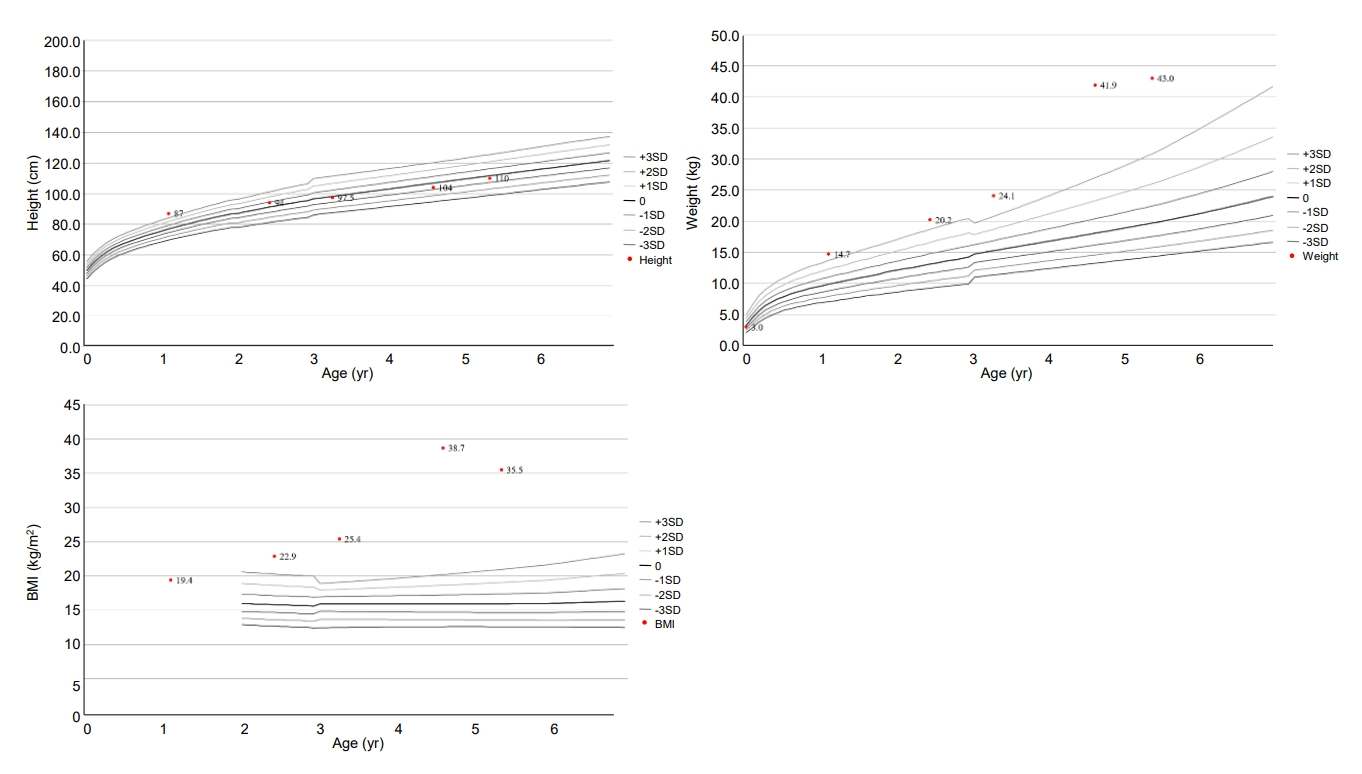
The patient was born at 37 weeks gestational age through in vitro fertilization with a birth weight of 3,000 g (z-score, -0.5 standard deviation score [SDS]). His body measurements, including birth height and head circumference, were within normal range. Meconium staining was not performed, and his vital signs were stable at birth. His APGAR score was 7 or higher. Laryngomalacia was observed until one month of age and improved naturally. There was no evidence of chorioamnionitis. Prenatal testing results, including amniocentesis, were normal. He was an only child and born to Korean non-consanguineous parents. His father had obesity, asthma, hypertension, and diabetes. Paternal height was 180 cm (1.2 SDS) and weight was 117 kg (4.2 SDS). Maternal height was 162 cm (0.1 SDS) and weight was 56 kg (-0.3 SDS). His grandparents had hypertension and diabetes. At 10 months old, he began toe walking, and his feet trembled when he was in a standing position due to spasticity of the lower limbs. He was diagnosed with cerebral palsy at the age of 13 months. At this point, height, weight, and body mass index (BMI) were 87.8 cm (2.9 SDS), 14.7 kg (3.5 SDS), and 18.8 kg/m2, respectively. Brain magnetic resonance imaging showed ventriculomegaly with prominent septum cavum pellucidum, which indicated periventricular leukomalacia. He primarily consumed formula until the age of 24 months. His height, weight, and BMI were 94 cm (0.9 SDS), 20.2 kg (4.2 SDS), and 22.7 kg/m2 (5.4 SDS) at 29 months old. He underwent rehabilitation treatment for cerebral palsy and nutritional counseling for obesity.
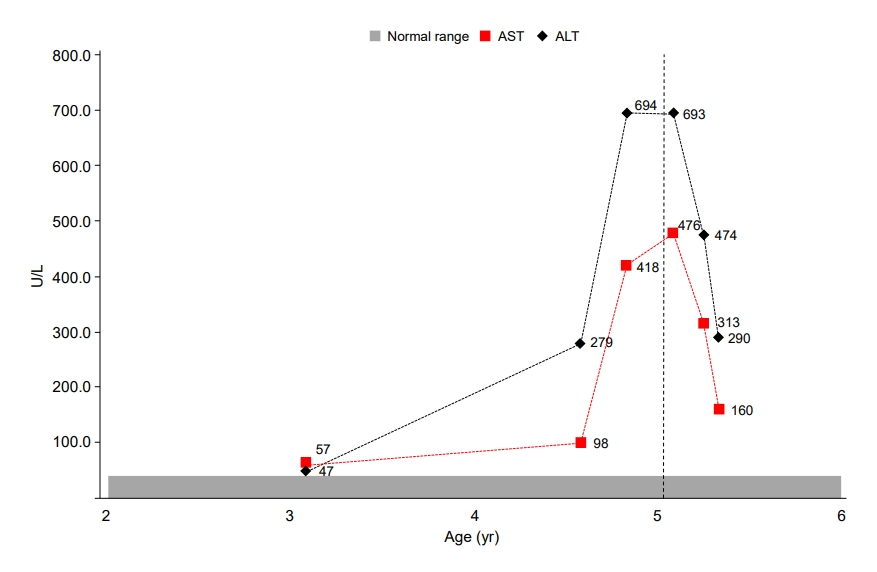
At the age of 4 years 7 months, the patient visited our department due to severe obesity. His height and weight were 104 cm (-0.5 SDS) and 41.9 kg (10.2 SDS), respectively, and his BMI was 38.7 kg/m2 (17.5 SDS) (Fig. 1). He had small hands and feet and showed severe truncal obesity. His cognition and language development were that of an 8ŌĆō10-month-old, and his motor development was that of a 10ŌĆō12-month-old. Laboratory tests revealed elevated aspartate aminotransferase (AST, 98 U/L; normal range, 0ŌĆō40 U/L); elevated alanine aminotransferase (279 U/L; normal range, 0ŌĆō41 U/L); elevated alkaline phosphatase (282 U/L; normal range, 40ŌĆō129 U/L); normal total bilirubin (0.2 mg/dL); normal total cholesterol (172 mg/dL); elevated triglycerides (181 mg/dL; normal range, 0ŌĆō149 mg/dL); normal high-density lipoprotein cholesterol (53 mg/dL), normal low-density lipoprotein cholesterol (99 mg/dL); normal glucose (96 mg/dL); elevated insulin (27.0 ╬╝IU/mL; normal range 1.1ŌĆō11.2 ╬╝IU/mL); and normal glycosylated hemoglobin (5.3%). His parents were aware of his elevated liver enzymes when they visited our clinic, and he had previously been diagnosed with NAFLD. He did not present with jaundice. Abdominal ultrasound revealed severe fatty liver. His viral and autoimmune hepatitis work-up was normal.
We attempted to find an underlying genetic defect to account for his early-onset severe obesity and associated elevated liver enzymes and developmental delays. Targeted gene panel sequencing was performed to detect the presence of genes potentially associated with early-onset severe obesity, hepatic disorders, and developmental delay, including associated diseases such as leptin receptor deficiency, POMC deficiency, and Alstr├Čm syndrome. The average read depth for each candidate gene is shown in Table 1. Informed consent was obtained from both parents. We obtained 3 mL of blood from the patient and both parents. Genomic DNA was extracted from the peripheral blood, and a cDNA library was prepared using the Celemics G-Mendeliome DES Panel (Celemics Inc., Seoul, Korea), which enriched a 12-Mb region spanning 70,418 target exons from a total of 5,447 clinically relevant genes. Massively parallel sequencing was performed on the Illumina NextSeq platform. Sequence reads were mapped to a UCSC hg19 standard base for conducting a comparative analysis. The average depth of the panel was 133.17X, and the percentage of bases above 10X of ATP7B was 100.0%.
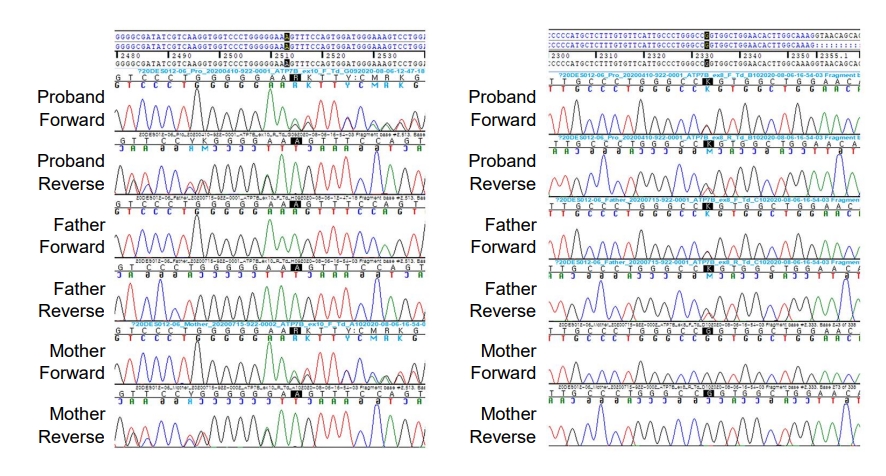
We found compound heterozygous mutations for c.2513del (p.Lys838SerfsTer*35) and c.2333G>T (p.Arg778Leu) in ATP7B on chromosome 13q14.3. Sanger sequencing confirmed the presence of these variants, and one of the same heterozygous variants was found in his father and mother, respectively (Fig. 2). These variants have been previously associated with WD. Serum ceruloplasmin declined to 4.09 mg/dL (normal range, 20ŌĆō46 mg/dL). Due to a lack of patient cooperation, a 24-hour urine specimen could not be collected and the Kayser-Fleischer ring in the cornea could not be examined. We started trientine (750 mg/day) to treat WD, and after 3 months of treatment, his liver enzyme began to decline (Fig. 3). Diet and exercise interventions for severe obesity were initiated.
Discussion
In this study, we report a male aged 4 years 7 months with obese WD accompanied by NAFLD. We diagnosed him with WD incidentally based on targeted gene panel sequencing to screen for genes related to pathologic obesity. Trientine was effective in improving his liver function.
NAFLD is associated with obesity. Numerous studies have indicated that NAFLD accounts for the hepatic component of metabolic syndrome characterized by obesity, insulin-resistance and type 2 diabetes, hypertriglyceridemia, and arterial hypertension. Although NAFLD was reported to be the most common liver pathology in WD patients, there are limited data on copper accumulation and NAFLD pathogenesis [3]. WD was first overlooked in our patient because of his morbid obesity and NAFLD. However, the most frequently observed hepatic histological lesion in WD is hepatic macro-steatosis and glycogenated nuclei, which are also features of NAFLD [2]. In a recently published study, hepatic steatosis in WD was not induced by metabolic comorbidities but by copper accumulation in liver tissue [3]. However, metabolic alterations could be cofactors in pathogenic steatosis in WD. Thus, NAFLD presence should not exclude a WD diagnosis.
Our patient was severely obese in early childhood. He typically enjoyed high-calorie foods, and his delayed motor development from cerebral palsy resulted in weight gain due to a lack of physical activity. A previous animal model study [4] suggested that WD may not be related to obesity. Atp7bŌłÆ/ŌłÆ mice were more glucose tolerant and insulin sensitive and showed decreased hepatic steatosis, while chow-fed Atp7bŌłÆ/ŌłÆ mice exhibited modest reduction in adiposity and body weight by 14 weeks of age compared with wild type mice. This suggests that obesity is less likely to develop in WD. Tribl et al. [5] also found that BMI was significantly lower in the WD group than in the control group in their case control study of sleep disorders in WD.
A few WD cases associated with other endocrinologic problems have been reported (Table 2). Growth and puberty may be affected by chronic systemic illness. A few cases of infertility, anovulation, amenorrhea, and short stature have been reported in patients with WD. Hypogonadotropic hypogonadism secondary to chronic liver disease is the most common cause of gonadal dysfunction in patients with WD [6,7]. One case showed growth hormone deficiency [8]. Kaushansky et al. [9] reported disturbed ovarian function in one WD family, as evidenced by low estradiol, high total testosterone, normal free testosterone, and elevated androstenedione. Another rare endocrinological disturbance is hypoparathyroidism, possibly due to copper accumulation in the parathyroid glands [10]. Krysiak et al. [11] reported 1 case of insulinoma and hyperprolactinoma in a female patient with WD, in which galactorrhea was the initial symptom 5 years before diagnosis. Golding et al. [12] and Canelas et al. [13] reported radiographic evidence of osteoporosis in up to 88% of people with WD.
This patient's neurological manifestations were developmental delay and seizure, both of which are not frequently reported in patients with WD. Periventricular leukomalacia is also not typically observed in WD. However, several studies have shown white matter lesions in patients with WD, suggesting association with copper accumulation. Assessment for improvements in developmental delay after trientine treatment might lead to a better understanding of its relevance in WD.
WD is controllable with appropriate copper chelating agents. In most patients with WD, early diagnosis and treatment generally result in symptom resolution and improvements in quality of life, growth problems, gonadal dysfunction, or other endocrinologic problems. Serum ceruloplasmin has been considered a simple method for testing WD. However, up to 20% of patients show normal or borderline ceruloplasmin level [14]. The 24-hour urinary copper excretion may be another useful tool for diagnosing WD and for monitoring treatment. However, ceruloplasmin and urinary copper levels are affected by acute infection, hepatic injury, renal failure, and certain medications such as steroids and estrogens. Moreover, Cauza et al. [15] found that the positive predictive value of low serum ceruloplasmin level was only 5.9%. These diagnostic tests are usually performed on those with newly onset hepatic abnormalities because serum AST generally increases in WD, except at very early disease stages. Only about 40% of pediatric patients present with hepatic symptoms [7]. In this respect, WD diagnosis could be challenging, especially in young patients or during the early disease phase.
Our patient was diagnosed with WD using a commercialized targeted gene panel that contained the WD-related gene. Although genetic testing for NAFLD in general cases is not yet recommended, the genetic etiology of NAFLD-related metabolic syndrome has been extensively studied. In WD, for nonspecific early symptoms such as NAFLD, generalizing next-generation sequencing in parallel with copper analysis is a promising step for early diagnosis. As genetic testing technology has been widely used, screening for asymptomatic families is gradually increasing, and the diagnosis age for WD has decreased to approximately 2ŌĆō3 years. Direct sequencing is a highly sensitive and widely used confirmation test method, and simultaneous biochemical testing improves its diagnostic accuracy. However, it is not widely available due to its cost.
In conclusion, increased liver enzymes are relatively common in obese children. A WD diagnosis should not be overlooked even in NAFLD cases that appear with obesity because WD can present as any liver disease.