Two cases of 17α-hydroxylase/17,20-lyase deficiency caused by the CYP17A1 mutation
Article information

Abstract
17α-hydroxylase/17,20-lyase deficiency, caused by mutations in the cytochrome P450 family 17 subfamily A member 1 gene (CYP17A1), is an extremely rare form of congenital adrenal hyperplasia that is characterized by diverse phenotypes resulting from specific mutations. Here, we report 2 phenotypic females with 17α-hydroxylase/17,20-lyase deficiency: one with the 46,XX karyotype presenting primary amenorrhea and sexual infantilism, and the other with the 46,XY karyotype presenting a disorder of sexual development. In both cases, the serum levels of adrenocorticotropic hormone, 11-deoxycorticosterone, and gonadotropin were elevated, whereas the levels of testosterone and dehydroepiandrosterone were reduced. Next-generation sequencing revealed one patient with compound heterozygosity for p.Trp17Ter (c.51G>A) and p.His373Leu (c.1118A>T), and the other with homozygosity for p.His373Leu (c.1118A>T). This report further describes 2 cases of 17α-hydroxylase/17,20-lyase deficiency in patients who harbored a p.His373Leu substitution, commonly found in Korean individuals, and presented diverse phenotypes.
Highlights
We introduce 2 cases of patients with 17α-hydroxylase/17,20-lyase deficiency. Both of them harbored a p.His373Leu substitution, providing an evidence of the founder effect of the p.His373Leu substitution in Korean patients with a 17α-hydroxylase/17,20-lyase deficiency.
Introduction
Congenital adrenal hyperplasia (CAH) is a group of inherited disorders caused by defects in enzymes involved in adrenal steroidogenesis, thereby resulting in various alterations in glucocorticoid, mineralocorticoid, and sex steroid production [1]. Among several genes causing CAH, cytochrome P450 family 17 subfamily A member 1 gene (CYP17A1) mutations lead to 17α-hydroxylase/17,20-lyase deficiency, which is an extremely rare form of CAH with an estimated worldwide incidence of 1 in 50,000 to 100,000 individuals [2]. 17α-hydroxylase/17,20-lyase are involved in androgen and cortisol synthesis from cholesterol; hence, their deficiencies cause androgen and cortisol deficiency, respectively. The diagnosis of a 17α-hydroxylase/17,20-lyase deficiency is quite challenging. This rare disorder differs from other forms of CAH, especially from 21-hydroxylase deficiency which accounts for approximately 95% of CAH cases and is characterized by an unelevated level of 17-hydroxyprogesterone (17-OH-progesterone). Furthermore, patients with 17α-hydroxylase/17,20-lyase deficiency usually have mild symptoms and signs of hypocortisolism, and they present primary amenorrhea and/or absence of secondary sexual characteristics during puberty [3]. Furthermore, phenotypes largely depend on whether these enzymes are completely or partially deficient. Impaired androgen production results in different phenotypes based on genetic sex: women with the 46,XX karyotype have normal female genitalia but diminished or obliterated sexual development at puberty, while those with the 46,XY karyotype have ambiguous or female external genitalia but no female internal genitalia. Therefore, patients present completely different clinical characteristics based on their karyotypes. Hence, various clinical symptoms should be meticulously assessed and thoroughly considered, and if doubtful, the diagnosis should be confirmed through genetic testing.
Here, we report 2 cases of 17α-hydroxylase/17,20-lyase deficiency in women, 1 with the 46,XX karyotype and 1 with the 46,XY karyotype, who harbored CYP17A1 mutations.
Case reports
1. Case 1
A 16-year-old female was transferred to the Department of Pediatric Endocrinology of Severance Children's Hospital for assessment of primary amenorrhea and features of sexual infantilism. The patient had no past medical or family history associated with disorders of sexual development. She was born at a gestational age of 41 weeks with a birth weight of 2.5 kg, compatible with small for gestational age. She was delivered through caesarean section and displayed no typical genital abnormalities postpartum. Initial endocrinological assessment, chromosomal analysis, and imaging investigation were carried out at another hospital. Initial endocrinological investigations revealed a hypergonadotropic hypogonadism with low estradiol and elevated follicle stimulating hormone (FSH) and luteinizing hormone (LH) levels. Decreased cortisol (1.1 µg/dL) and elevated adrenocorticotropic hormone (ACTH) (181.8 pg/mL) levels were noted. The patient's karyotype was 46, XX, and pelvic ultrasonography and pelvic magnetic resonance imaging revealed a small uterus measuring 2 cm without visible endometrial tissue, a normal vagina, and ovaries with multiple cystic lesions.
She was referred to our endocrinology clinic for further evaluation of primary amenorrhea with primary gonadal failure and suspected hypocortisolism. Physical examination revealed a normal height and weight of 158.3 cm (25th–50th percentile) and 45.3 kg (5th–10th percentile), respectively. Her breasts were at Tanner stage II, and pubic hair was at Tanner stage I. No skin pigmentation was observed. Her blood pressure was in the upper normal limit at 129/99 mm Hg (90th–95th percentile). Laboratory investigations revealed normal electrolytes and thyroid function, hypocortisolism (cortisol, 1.9 µg/dL; ACTH, 1,815 pg/mL), and hypergonadotropic hypogonadism (estradiol, <8 pg/ml; LH, 47.2 mIU/mL; FSH, 18.2 mIU/mL). Meanwhile, she presented a normal 17-OH-progesterone (0.91 ng/mL) and elevated 11-deoxycorticosterone level (12.30 ng/mL; reference range, 0.02–0.19 ng/mL). The aldosterone-to-renin ratio was 39.4 (cutoff, <23.6) with elevated aldosterone levels (209 pg/mL; reference range, 10–160 pg/mL) and low plasma renin activity (PRA) (0.53 ng/mL/hr; reference range, 1.31–3.95 ng/mL/hr), indicating hyperaldosteronism (Table 1). After the patient was stimulated with synthetic ACTH, the levels of cortisol, 17-OH-progesterone, 17-hydroxypregnenolone (17-OH-pregnenolone) and dehydroepiandrosterone (DHEA) remained unchanged (Table 1). Based on these results, a 17α-hydroxylase deficiency was suspected, and genetic testing was performed for CYP17A1. Clinical exome sequencing was performed using the TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA, USA), which enriches a 12-Mb region spanning 62,000 target exons of a total 4,813 genes. Compound heterozygous mutations for p.Trp17Ter (c.51G>A) and p.His373Leu (c.1118A>T) were identified in CYP17A1 (Fig. 1A). As both mutations were evaluated as pathogenic according to the 2015 ACMG-AMP guidelines [4], the diagnosis of a 17α-hydroxylase/17,20-lyase deficiency was confirmed.
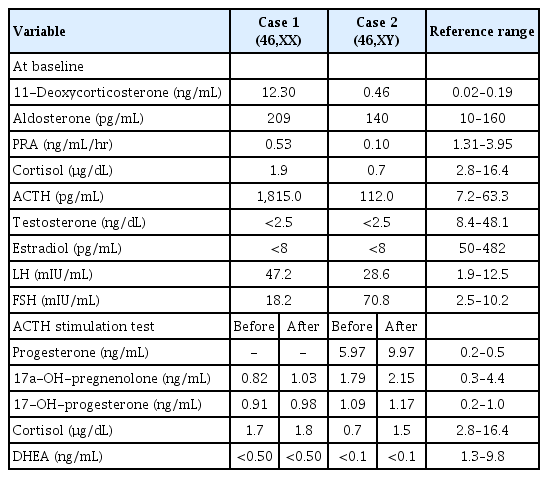
Hormonal findings at baseline and the results of the synthetic ACTH stimulation test in cases 1 and 2

(A) Direct sequencing of the CYP17A1 gene from the patient in case 1. Heterozygous p.Trp17Ter (c.51G>A), p.His373Leu (c,1118A>T) mutations were identified. (B) Next-generation sequencing for hypogonadism from the patient in case 2. A homozygous p.His373Leu (c.1119A>T) CYP17A1 mutation was identified. P, pathogenic; U, uncertain significance.
After receiving the diagnosis of a 17α-hydroxylase/17,20-lyase deficiency, the patient started oral hydrocortisone treatment (10 mg/m2/day). Furthermore, Femoston, a combination of 2-mg estradiol and 10-mg progesterone, was initiated. Four months after treatment initiation, physical examination revealed Tanner stage III breasts and Tanner stage II pubic hair, her blood pressure decreased to 110/70 mm Hg (25th–50th percentile), and she began menstruation. Laboratory tests revealed normal levels of cortisol (6.9 µg/dL), ACTH (5.9 pg/mL), estradiol (90.84 pg/mL), LH (6.16 mIU/mL), and FSH (8.3 mIU/mL) at a 6-month follow-up evaluation. Furthermore, transabdominal sonography 6 months after treatment revealed a normal uterus measuring 15.9 mL (normal range, 21.2±13.5 mL).
2. Case 2
A 4-year-old phenotypic female patient who presented with a bulging right inguinal area underwent right hernioplasty. As the hernia sac displayed prepubertal testis without ovarian stroma, she was referred to our endocrinology clinic for further evaluation of disorders of sexual development. She was born at full-term through caesarean section, and her birth weight was 3.41 kg. She did not have a specific medical or family history. Physical examination revealed normal female external genitalia with a normal clitoris, urethra, and vaginal opening. Her growth status was normal, with a height and weight of 115 cm and 23.9 kg (90th–95th percentile in age-matched girl), respectively. Her blood pressure was 145/100 mm Hg (>97th percentile). Initial endocrinological investigations were as follows: LH, 28.6 mIU/mL; FSH, 70.8 mIU/mL; testosterone, <2.5 ng/dL; estradiol, <8 pg/mL; cortisol, 0.7 µg/dL (reference range, 2.8–16.4 µg/dL); ACTH, 112.0 pg/mL (reference range, 7.2–63.3 pg/mL); serum aldosterone, 140 pg/mL (reference range, 10–160 pg/mL); and PRA, 0.10 ng/mL/hr (reference range, 1.31–3.95 ng/mL/hr). The findings indicated mild hypocortisolism and elevated gonadotropin (Table 1). Meanwhile, she presented a normal 17-OH-progesterone (1.09 ng/mL) and elevated progesterone (5.97 ng/mL; reference range, 0.2–0.5 ng/mL) and 11-deoxycorticosterone level (0.46 ng/mL; reference range, 0.02–0.19 ng/mL). The aldosterone-to-renin ratio was 140, indicating hyperaldosteronism, although an electrolyte imbalance was not observed. A high-dose synthetic ACTH stimulation test revealed no response of cortisol, 17-OH-progesterone, 17-OH-pregnenolone, and DHEA, but rather revealed an elevated progesterone response (from 5.97 to 9.97 ng/mL) (Table 1). Chromosomal analysis and fluorescence in situ hybridization revealed a normal male karyotype, 46,XY, with SRY. Pelvic ultrasonography revealed no visible uterus or ovaries in the pelvic cavity, and genitography revealed a normal urethral and vaginal opening. With the suspicion of 17α-hydroxylase/17,20-lyase deficiency, we performed next-generation sequencing-based targeted gene panel for hypogonadism that encompassed 36 related genes (AKAP6, AMH, CDKL5, CGA, CRK, CYP17A1, CYP19A1, CYP1A1, DLK1, ESR1, FSHR, GH1, GHRH, GNAS, HRAS, IGF1, IGFALS, INS, KISS1, KISS1R, LEP, LEPQTL1, LEPR, LHB, LHCGR, LIN28B, MKRN3, NF1, NFIX, NNT, NOTCH1, NR0B1, OGDHL, PROKR2, PTPN11, and STK11) using Miseq (Illumina, Inc.). A homozygous p.His373Leu (c.1118A>T) mutation of the CYP17A1 gene was identified, confirming 17α-hydroxylase/17,20-lyase deficiency (Fig. 1B).
Oral hydrocortisone (12 mg/m2/day) treatment was initiated. Given her clinical features, sex of rearing, and parents’ opinion, the patient was raised as a girl, and she underwent orchiectomy on the opposite side. Pathological assessment revealed a prepubertal testis with no evidence of residual germ cells and slightly increased Leydig cells. After treatment with hydrocortisone, the patient's blood pressure and serum levels of cortisol and ACTH remained within normal ranges. Low-dose oral estradiol treatment was initiated at 12 years of age to promote the development of secondary sexual characteristics.
Discussion
17α-hydroxylase/17,20-lyase deficiency is a rare form of CAH, accounting for only approximately 1% of all CAH cases [2,5]. The first reported case was a 35-year-old patient in San Francisco in 1966 who was both genotypically and phenotypically female but presented with sexual infantilism and primary amenorrhea [6]. As patients with this disorder can present various phenotypes based on the types of mutations, genetic analysis is necessary. Hence, a thorough pathophysiological understanding of the clinical manifestations and molecular derangements is required. CYP17A1 (10q24.3) encodes the P450c17 enzyme, a type 2 microsomal P450 enzyme expressed in the adrenal gland and gonads, which performs both steroid 17α-hydroxylase and 17,20-lyase activities [5,7]. Both enzymes contribute to cortisol and sex steroid production through the steroid biosynthesis pathway. 17α-hydroxylase converts pregnenolone to 17-OH-pregnenolone and progesterone to 17-OH-progesterone, and 17,20-lyase further converts to DHEA and androstenedione. Defects in 17α-hydroxylase/17,20-lyase result in a reduction in the 17-OH-pregnenolone and 17-OH-progesterone levels, resulting in low cortisol levels and a decline in DHEA and androstenedione, which in turn result in low adrenal androgen levels. Low cortisol levels result in the overstimulation of the steroid synthetic pathway as negative feedback, resulting in ACTH overproduction and adrenal cortex hyperplasia. Since CYP17A1 is expressed in the zona reticularis and zona fasciculata of the adrenal gland, but not in the zona glomerulosa, steroidogenesis through ACTH stimulation leads to deoxycorticosterone and corticosterone overproduction [3]. Elevated deoxycorticosterone levels lead to sodium retention, hypokalemia, hypertension, and suppression of aldosterone production. Although cortisol production is low or absent in 17α-hydroxylase/17,20-lyase deficiency, adrenal disorders seldom occur owing to the presence of corticosterone, which has glucocorticoid activity [3].
As of September 2020, 123 CYP17A1 mutations have been identified in the Human Gene Mutation Database. The first mutation was reported in a Thai patient of Chinese descent as a 4-base insertion in 1993 [8]. Different populations harbor different mutations with regional accumulation as a founder effect. Since the first report of a homozygous missense p.His373Leu substitution in 2 Japanese sisters in 1993 [9], the p.His373Leu mutation has been frequently reported in Japanese, Chinese, and Korean individuals, which is presumed to exert a founder effect in Northeast Asian populations [10,11]. Since the first p.His373Leu mutation in a Korean individual was reported in 2012, 10 cases, including the present 2 patients, have been reported to carry the CYP17A1 mutations with the p.His373Leu substitution in the Korean population [10,12,13]. These Korean cases provide robust evidence that the p.His373Leu substitution exerts a founder effect, especially in Korean patients with a 17α-hydroxylase/17,20-lyase deficiency. The previous functional study revealed that COS-1 cells transfected with a p.His373Leu mutant obliterate both 17α-hydroxylase and 17,20-lyase activities [9]. It has been proposed that p.His373Leu substitution does not allow for appropriate incorporation of the heme moiety, leading to a global alteration of P450c17 structure and consequently prevents heme binding, thus attenuating enzyme activity [9,14]. Furthermore, a previous study reported that the p.His373Leu mutant did not have 11β-hydroxylase and aldosterone synthase activities since the mutant failed to convert [14C]deoxycorticosterone to corticosterone or aldosterone [9]. Most cases of 17α-hydroxylase/17,20-lyase deficiency revealed low aldosterone levels owing to suppression of the renin-angiotensin system. However, our patients displayed discrepancies, revealing high aldosterone levels with suppressed PRA, similar with previous reports [9,12]. While the reason underlying this difference remains unclear, it is speculated that the presence of low-level enzyme activity based on the genotype or unknown factors contribute to phenotypic variation [15]. Furthermore, Monno et al. reported that aldosterone secretion may be influenced by an increase in ACTH levels and not by the activation of the renin-angiotensin system [9]. Like glucocorticoid-remediable aldosteronism, which has a chimeric 11β-hydroxylase/aldosterone synthase carrying out aldosterone synthesis with cortisol and cortisol precursors [16], plasma aldosterone levels in patients with 17α-hydroxylase/17,20-lyase deficiency appear to be regulated by ACTH.
In conclusion, this case report describes 2 additional cases of CYP17A1 mutations in Korea, both harboring a p.His373Leu substitution. One harbored a heterozygous mutation, and the other harbored a homozygous mutation. It may be speculated that the founder effect of the p.His373Leu substitution manifests among Korean individuals harboring CYP17A1 mutations. Because patients with 17α-hydroxylase/17,20-lyase deficiency present variable clinical characteristics based on their karyotypes, meticulous assessment including genetic testing is necessary.
Notes
Ethical statement
This case was approved by the Institutional Review Board of Yonsei University Health System, Severance Hospital (approval number: 4-2020-0604). Informed consents were obtained from the patients and their parents.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.