Highlights
· Male sexual development requires the action of testicular testosterone and dihydrotestosterone (DHT). DHT is the most active endogenous androgen, and it is converted from testosterone in genital skin. This route from cholesterol to DHT is called the conventional classic pathway. Recent reports have revealed a backdoor route to DHT that circumvents testosterone production. Androsterone is the primary circulating backdoor androgen. Furthermore, androsterone and steroidal substrates specific to the backdoor route are predominantly found in the placenta, liver, and adrenal glands rather than the testes. These findings are crucial to our understanding of human sexual development.
Introduction
Sex development mainly occurs during the first half of gestation, with the end form of sexual structures dependent on sex chromosomes. 46,XX chromosomes in addition to genetic factors like DAX1 (dosage-sensitive/sex-reversal adrenal hypoplasia on the X chromosome), WNT-4, and RSPO1 are required for the development of ovaries [1,2]. However, the development of the male sex phenotype is more complex. 46,XY chromosomes with an intact SRY (sex-determining region on the Y chromosome) gene, as well as transcription factors like SOX9, SF-1 (steroidogenic factor-1), and WT1 (Wilms tumor 1), are needed for testicular development [1,2]. Fetal testicular Sertoli cells produce anti-Müllerian hormone (AMH), which is responsible for regression of Müllerian duct derivatives, including the uterus, fallopian tubes, cervix, and upper vagina. AMH activation is most likely mediated by the SF-1 gene [1]. By approximately 8 weeks of gestation, Leydig cells produce testosterone from cholesterol, which is sequentially mediated by several enzymes. Subsequently, testosterone facilitates the development of the Wolffian duct into the epididymis, vas deferens, and seminal vesicles. Complete development of external genitalia requires the action of dihydrotestosterone (DHT), the most potent testosterone metabolite. DHT is essential for fusing genital folds to form the penis and scrotum [2]. In genital skin, testosterone undergoes conversion into DHT via the action of 5α-reductase type 2, which is encoded by the SRD5A2 gene [3,4]. This process is called the conventional classic pathway. Recent studies revealed that DHT is also generated via an alternative ("backdoor") route from androstanediol, and this route must be intact for normal and complete prenatal masculinization [5-9]. In this review, we highlight new evidence clarifying this backdoor pathway.
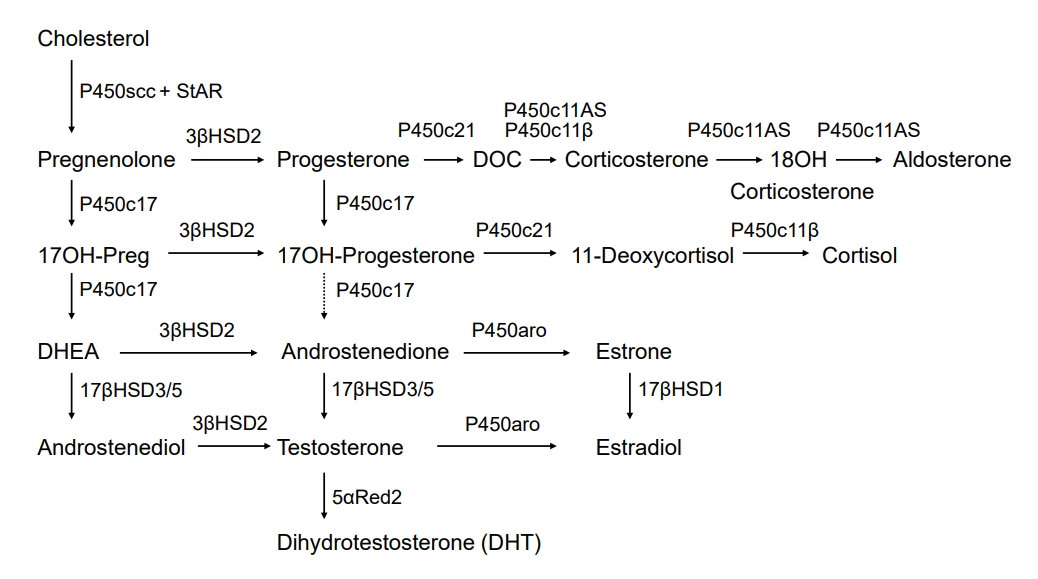
Classic pathway of androgen synthesis
The classic route of androgen biosynthesis, both in the testis and adrenal zona reticularis, is well established [10]. Cholesterol is the precursor for androgen biosynthesis, and cholesterol transformation to pregnenolone is the initial rate-limiting step [11]. The steroidogenic acute regulatory protein (StAR) initiates the transport of cholesterol into the mitochondria [12]. Removal of the cholesterol side-chain occurs in the inner mitochondrial membrane, producing pregnenolone; this step is catalyzed by a cholesterol side-chain cleavage enzyme (P450scc; previously termed 20,22 desmolase), which is a cytochrome P450 (CYP) enzyme [13]. P450scc engages cholesterol 20α-hydroxylation, 22-hydroxylation, and side-chain cleavage to produce pregnenolone. Three distinct enzymes were originally thought to mediate these reactions, but P450scc, encoded by the CYP11A1 gene on chromosome 15, was found to catalyze all the steps that mediate pregnenolone formation from cholesterol. P450scc receives electrons from a reduced nicotinamide adenine dinucleotide phosphate-dependent mitochondrial electron transport system consisting of 2 accessory proteins, ferredoxin (also termed adrenodoxin) and ferredoxin reductase (adrenodoxin reductase) [13]. P450scc can only work within mitochondria; hence, cholesterol transfer to the inner mitochondrial membrane by StAR is a critical step for androgen synthesis [13]. The zona reticularis and, to some extent, the zona fasciculata, express both 17α-hydroxylase and 17, 20 lyase activities of P450c17, thereby yielding dehydroepiandrosterone (DHEA) [10,14]. The 17, 20 lyase activity of P450c17 requires allosteric action of cytochrome b5 in the adrenal zona reticularis and testicular Leydig cells. Human P450c17 catalyzes 17OH-progesterone to androstenedione with only 2%–3% of 17OH-pregnenolone transformed into DHEA, so testosterone production occurs through DHEA and not through 17OH-progesterone; however, rodent and ungulate P450c17 can effectively mediate this reaction (Fig. 1) [10]. In this process, P450c17 is supported by cytochrome P450 oxidoreductase (POR) for electron transfer [10,15]. DHEA is transformed into androstenedione by 3β-hydroxysteroid dehydrogenase type 2 in both the adrenal glands and testes; however, the testes continue to be the primary site for the following androstenedione transformation into testosterone by 17β-hydroxysteroid dehydrogenase type 3 (17βHSD3). Low levels of adrenal 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5, also known as AKR1C3) can induce the production of small amounts of testosterone [10]. The resulting testosterone is transported into circulation and absorbed by the genital skin, where it is transformed into DHT by 5α-reductase type 2 (Fig. 1) [3,4].
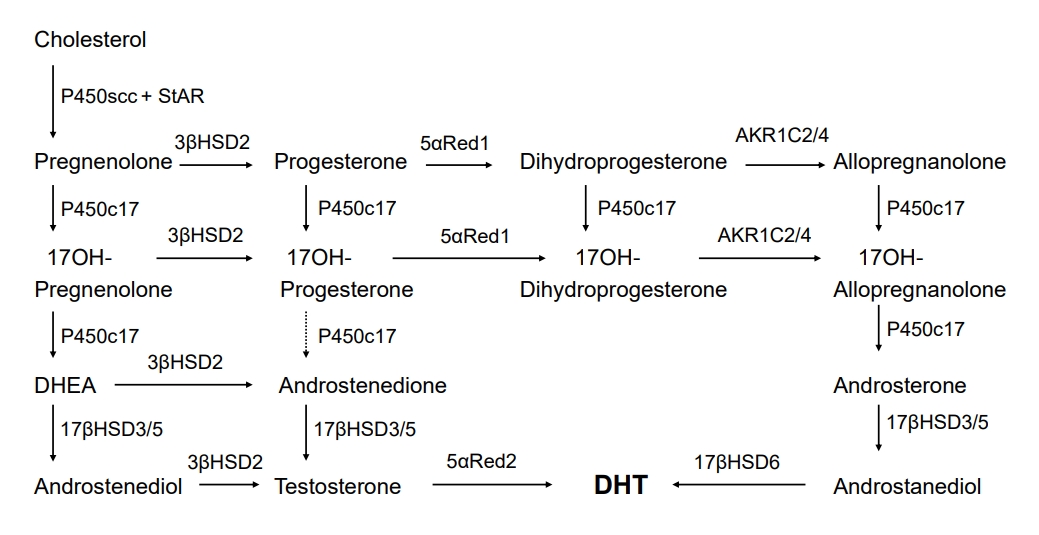
Backdoor pathway of androgen synthesis
An alternative route of androgen synthesis has been studied in the gonads of the tammar wallaby and immature mouse testes [16-18]. The tammar wallaby, a small marsupial mammal, is an advantageous model for investigating male phenotypic development, as male genital development occurs over a prolonged period postnatally in the mother's pouch, allowing easy access for tissue sampling without placental intervention [19]. Research using young tammar wallabies revealed an alternative route for DHT production. This route circumvents conventional intermediate steroids such as DHEA, androstenedione, and testosterone. Alternatively, 17OH-progesterone can be produced from 17OH-pregnenolone, with the subsequent reduction of 5α- and 3α-, subject to the 17, 20 lyase activity of P450c17, followed by 17βHSD3-mediated catalysis to form androstanediol as the circulating product [17]. As observed in eutherian mammals, target tissues carry out the final enzymatic conversion to generate DHT [17]. In addition, similar reports in immature mouse testis have revealed 2 different routes for DHT formation; the classic route mediated by testosterone and the backdoor route mediated by androstanediol, generated from progesterone through dihydroprogesterone (5α-pregnane-3,20-dione), allopregnanolone (5α-pregnane-3α-ol-20-one), 17OH-allopregnanolone, and androsterone [18]. These studies revealed the existence of a pathway sequentially converting cholesterol into DHT without the requirement for testosterone [20]. The backdoor route is controlled by several enzymes. First, 5α-reductase is important for this pathway [10,20,21]. Both progesterone and 17OH-progesterone are precursors for 5α-reductases, mainly 5α-reductase type 1 (SRD5A1) [22]. which is expressed in human fetal testes [6]. Second, P450c17, which is involved in the classic pathway, is also essential to the backdoor route. Dihydroprogesterone and allopregnanolone are precursors for the 17α-hydroxylase of P450c17, and 17OH-allopregnanolone is the most effective precursor for 17, 20 lyase [10,23]. In addition, other enzymes like AKR1C2/4, 17βHSD5 (AKR1C3), and 17βHSD6 (also called retinol dehydrogenase, RoDH) are critical mediators of this pathway (Fig. 2) [6]. Various factors determine the predominance of the classic or backdoor routes. The most significant factor is P450c17 when compared with 5α-reductase type 1, given that this activity determines whether 17OH-progesterone and progesterone are utilized in the classic or backdoor pathway [20,21]. The functional level of 17, 20 lyase is also an important factor. The 17, 20 lyase activity of P450c17 was proportionally low in tammar wallabies, resulting in elevated 17OH-progesterone levels in the classic route, thereby inducing entry into the backdoor route [17,20].
Backdoor route in male sexual development
The human backdoor route has been implicated as an essential route of androgen synthesis for male genital development. This alternative pathway plays a crucial role in human hyperandrogenic disorders like congenital adrenal hyperplasia caused by P450c21 deficiency [8], polycystic ovary syndrome [24], and POR deficiency [25-27], and in physiological male minipuberty during infancy [28]. In addition, mutations in AKR1C2 and AKR1C4, the genes encoding 3α-reductases (3αHSD), reportedly cause disorders of sexual development (DSD), suggesting that both the classic and backdoor routes are required for normal human male sexual development [6,29].
1. Backdoor pathway in P450c21 deficiency
Congenital adrenal hyperplasia is an autosomal recessive disorder. P450c21 (21-hydroxylase) deficiency reportedly underlies >90% of cases of congenital adrenal hyperplasia, owing to mutations in the CYP21A2 gene [30,31]. This enzyme transforms progesterone into deoxycorticosterone and 17OH-progesterone into 11-deoxycortisol. Female infants with classic P450c21 deficiency may present with abnormal development of external genitalia along with varying degrees of masculinization [32,33]. With P450c21 deficiency, androgens are produced via all three routes [34]. First, the conventional route from cholesterol to DHEA remains intact; although a considerable amount of DHEA is inactivated to DHEA sulfate, the increased DHEA yield will induce some DHEA to undergo transformation into testosterone and DHT [35]. Second, while trivial amounts of 17OH-progesterone are transformed to androstenedione in the normal adrenal gland, the considerable 17OH-progesterone levels generated in P450c21 deficiency induce 17OH-progesterone transformation into androstenedione and subsequently into testosterone [35]. As 17βHSD5 (AKR1C3) enzyme is present in the fetal adrenal gland, it can induce the transformation of some androstenedione into testosterone, predominantly throughout the fetal period via the interrelationship between DHEA transformation into androstenedione and androstenedione transformation into testosterone [36,37]. Third, the backdoor route is reportedly involved in the masculinization of female patients with P450c21 deficiency, which is associated with elevated 17OH-progesterone entering the backdoor route in the adrenal gland. This alternate route relies on the 5α and 3α reduction of 17-OH progesterone to 17OH-allopregnanolone; this steroid is readily transformed to androstanediol, which can then be oxidized to DHT by an oxidative 17βHSD6 (RoDH) enzyme (Fig. 2). This backdoor route is evidenced by elevated levels of unique steroidal intermediate metabolites in the urine of infants, children, and adults with P450c21 deficiency [8]. Kamrath et al. [8] studied urine steroid profiles in 142 individuals with P450c21 deficiency and revealed notably elevated concentrations of 17OH-allopregnanolone, the main steroidal intermediate of the backdoor route. The authors also presented a higher androsterone to etiocholanolone (a metabolite of DHEA) ratio in individuals with P450c21 deficiency than in controls. Androsterone can not only originate from the conventional route but also the backdoor route [17,18,20,23,26,38]. However, etiocholanolone is produced most only via the conventional route [26]. Accordingly, the ratio of androsterone to etiocholanolone acts as a barometer for the amount of androsterone originating from the alternative backdoor route [26].
2. Backdoor route in POR deficiency
The alternative backdoor route was suggested following a urinary steroid metabolite study in POR deficiency [26]. POR deficiency is an autosomal recessive disease occurring as a result of mutations in the POR gene, which encodes electron transfer to microsomal enzymes involving P450c17, P450c21, and P450aro [39,40]. POR deficiency presents diverse clinical phenotypes, including skeletal defects and adrenal steroidogenic impairment with elevated 17OH-progesterone, undervirilization in 46,XY patients, and masculinization of 46,XX patients [39]. While it remains perplexing that affected females could be masculinized despite incomplete P450c17 function (which is needed for androgen synthesis), an alternative backdoor route reportedly exists in which 17OH-progesterone undergoes transformation to 17OH-allopregnanolone, a more useful substrate for the 17, 20 lyase activity of P450c17 than the conventional substrate, 17OH-pregnenolone [41]. Subsequently, 17OH-allopregnanolone is transformed via several enzymatic steps to DHT, the most active androgen [26]. Furthermore, the function of P450aro in the placenta is reduced, resulting in the unimpeded action of androgens generated by the fetal adrenals. This, in turn, aggravates female fetus masculinization and might also induce the masculinization of women pregnant with an affected fetus. This has been confirmed by decreased estriol levels found in women pregnant with a fetus exhibiting POR deficiency [27]. Patients with POR deficiency excrete urinary steroid substances, suggesting the involvement of the backdoor pathway [26,41-44]. In patients with POR deficiency, pregnanediol (a metabolite of progesterone), pregnanetriolone (a metabolite of 21-deoxycortisol), and pregnanetriol (a metabolite of 17OH-progesterone) were notably increased, and urine steroid ratios showing P450c17 and P450c21 activity were decreased [26,41]. Moreover, etiocholanolone (a metabolite of DHEA) and 11-hydroxyandrosterone, associated with the classic pathway, exhibited normal or slightly decreased levels; however, androsterone, which is associated with both pathways, was elevated. This indicates the existence of the backdoor route in POR deficiency [26].
3. Backdoor pathway in DSD
Mutations causing DSD have been shown in AKR1C2 and AKR1C4 genes encoding 3α-reductases (3αHSD), which function exclusively in the backdoor pathway (Fig. 2) [6]. AKR1C2 is an enzyme involved in the backdoor pathway but not the conventional classic pathway. Fluck et al. [6] reported AKR1C2 gene mutations with X-linked recessive inheritance in patients with 46,XY DSD. These patients presented with moderate to severe undermasculinization at birth. Patients with 46,XY DSD also exhibit a mutation that causes abnormal splicing in the AKR1C4 gene, encoding an enzyme with similar activity. An individual with 46,XY DSD was found to exhibit AKR1C2 gene mutations on both alleles, supporting the crucial role of AKR1C2 [6]. This report revealed that both the classic and backdoor pathways are required for normal human male sexual development [6,29].
4. Major human backdoor androgen
Previous works on the backdoor pathway have only provided indirect evidence, as circulating and tissue androgen levels were not determined in examined fetuses. O'Shaughnessy et al. [7] measured plasma and tissue levels of steroids implicated in the classic and backdoor routes in second-trimester human fetuses using multidimensional and high-resolution mass spectrometry. The study revealed that androsterone, rather than testosterone, is the main backdoor androgen in male fetal circulation; however, significantly lower androsterone and testosterone concentrations were detected in female fetuses. In males, steroidal substrates specific to the backdoor route were predominant in the placenta and fetal liver, with significant androsterone concentrations detected in the fetal adrenal gland. Backdoor substrates, including androsterone, were only present at markedly low concentrations in the fetal testes. A PCR study revealed similar distributions of P450c17, 5α-reductase type 2, and AKR1C2/4 mRNA expression levels in the same tissues, indicating the generation of these steroids within these tissues [7]. These findings indicated that androsterone is the principal backdoor androgen in the human fetus and revealed that circulating concentrations are sex-dependent; however, small amounts of de novo synthesis occur in the testis. Also, data suggest that placental progesterone acts as a substrate to produce backdoor androgens, which reportedly occurs across several tissues. Accordingly, virilization of the human male fetus is mediated via testosterone and androsterone production by both the fetal testes and nongonadal tissues, resulting in DHT synthesis in genital skin [5,7].
Conclusions
DHT is the most active endogenous androgen, and is generated from testosterone in genital skin. This DHT synthesis route, cholesterol to DHT generation, is called the conventional classic pathway. The human fetal testis is essential but insufficient to generate androgenic steroids responsible for male genital development. Recent investigations have suggested that the backdoor route generating DHT can circumvent testosterone production. The backdoor route appears to participate in normal physiological virilization, as well as in abnormal masculinization during pathological states. It has been suggested that androsterone is the primary androgen of the human backdoor route, which reportedly originates from placental progesterone metabolized to androsterone in nontesticular tissues. However, some characteristics of the human backdoor pathway need to be further clarified. Future investigations require to focus on elucidating steroidal pathways in various tissues mediated via several steroidogenic steps.