Introduction
Short stature (SS) is the most common cause of referral to pediatric endocrinologists [1]. Only a minority of children with SS, which can be defined as a height less than 2 standard deviation score (SDS) below the mean, have an underlying pathology [2]. Although wellestablished diagnostic and management paradigms do exist, recent advances in molecular technologies have significantly improved our understanding of the genetic causes of SS, resulting in modification of treatment of children with different pathologies. In particular, although assessment of the growth hormone (GH)-insulin-like growth factor-1 (IGF-1) axis is often part of the early clinical evaluation of growth problems in children, our evolving understanding of growth plate physiology has led to a shift in focus on defects in this tissue, resulting in potential targets for the development of future innovative treatments [3].
Guidelines for referral vary widely among countries, but early diagnosis and initiation of treatment are critical, particularly in children who are candidates for GH treatment [4]. However, GH concentrations vary depending on the immunoassays used [5,6], age and body mass index (BMI) of the patient [7,8], and cutoff values of the GH provocation tests used to determine if GH treatment of a patient is warranted.
This review summarizes some of the key findings in children with SS and focuses on some of the indicators of the underlying pathology as well as potential future treatment options.
Physiopathology of growth
1. GH and the IGF‐1 system
The GH-IGF-1 axis is the most important endocrine system regulating linear growth during childhood. [9] In association with nutritional status, GH is a relevant stimulator of IGF-1 secretion in children and adolescents.
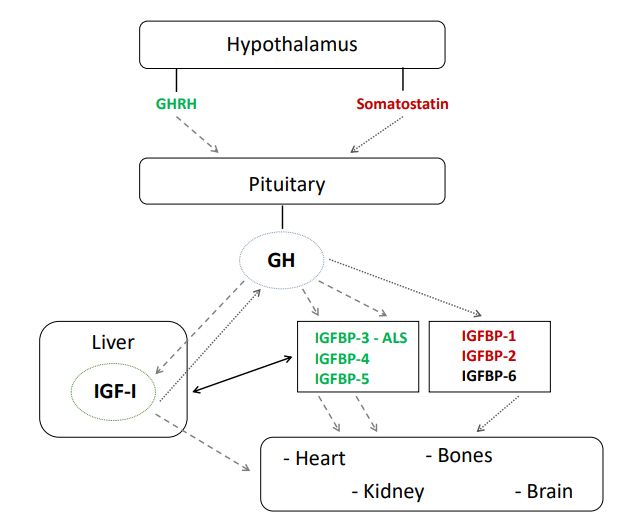
GH is a 191 amino acid protein (molecular weight of 22 kDa) produced and released in a pulsatile fashion from the anterior pituitary under positive control of GH-releasing hormone and negative control of somatostatin, both produced by the hypothalamus [10]. GH acts on the liver to determine the release of both glucose and IGF-1, as well as in on adipose tissue, where it modulates the concentration of fatty acids [11]. Furthermore, GH acts on the cartilage cells of the growth plates, resulting in IGF-1 secretion that modulates bone length during childhood [3].
IGF-1 itself is a small protein of 70 amino acids (molecular weight of 7.65 kDa) that is produced by many cells, in particular hepatocytes. Specific insulin-like growth factor-binding proteins (IGFBPs) bind IGF-1 in the circulatory system. GH inhibits production of IGFBP-1 and -2, but induces production of IGFBP-3, -4, and -5 [12]. IGFBP-3 is produced by hepatic sinusoidal cells at the junction of the intravascular space. IGF-1 is mainly bound to IGFBP-3, which then binds to a large protein named acid-labile subunit (ALS) to form a ternary complex [9]; this complex inhibits both IGF-1 and IGFBP-3 production and extends their half-life in the circulation. When IGF-I is bound to IGFBP-3, it is not active, but can be released in peripheral tissues. Free IGF-1 then interacts with cell surface receptors to activate a signaling pathway within the cells (Fig. 1). In addition, IGFBP-3 can also bind to a specific receptor that then translocates to the cell nucleus where it interacts with the retinoid X receptor and nuclear receptor to form a complex that can modulate transcription and induce apoptosis [13].
GH is the most important stimulator of IGF-1 and IGFBP-3 secretion from the liver [14]. However, many other hormonal factors control IGF-1 concentration in the circulation such as insulin [15], thyroid hormone [16], and androgens. Interestingly, estrogens at low levels stimulate the secretion of IGF-1 while at high concentrations, they reduce its secretion [17].
Malnutrition and other conditions such as celiac disease [18] and anorexia [19] can reduce serum concentrations of IGF-1, IGFBP-3, and ALS. In particular, chronic inflammation such as that associated with Crohn disease or juvenile chronic arthritis can interfere with the normal function of GH and IGF-1 (this is known as the 'dual-effector theory'). Indeed, pro-inflammatory cytokines, such as tumor necrosis factor alpha and interleukin 6 may interfere with systemic release of IGF-1, determining hepatic GH resistance [20]; in addition, they may inhibit the action of locally produced IGF-1, resulting in reduced proliferation and differentiation of chondrocytes in the growth plate [21].
2. Growth plate composition and hormonal regulators
A network of signals influences bone growth [3]. Both GH and IGF-1 are part of this network and modulate chondrocyte proliferation and differentiation. Cartilage growth plates contain 3 main layers known as the resting zone, proliferative zone, and hypertrophic zone. Each layer is distinguished by different rates of proliferation and differentiation of chondrocytes, which are the only cell type in this tissue [22]. GH has been reported to act in the resting zone to induce chondrocytes to differentiate and proliferate [23]. IGF-1 could determine cellular differentiation in the proliferative and hypertrophic zones, thus increasing height within columns of chondrocytes that then calcify the surrounding matrix [24].
Normal growth
A normal growth trajectory typically indicates good general health, while growth that is slower than normal may suggest an underlying pathologic condition, including hormonal causes of growth failure [25].
Growth is a continuous, but not linear, process; as is well known, children have 3 different phases of growth: infantile, childhood, and pubertal. These phases are similar for boys and girls, but the timing and place of growth vary, particularly during puberty. Size at birth is influenced more by maternal diet and intrauterine and placental elements than by genetic factors. Not all the genes that modulate growth are expressed at birth. Therefore, there may not be a correlation between length at birth and adult height. In the first 2 years of life (infancy growth phase), linear growth is very rapid but decreases thereafter; the overall growth is approximately 30 to 35 cm. An infant's height curve frequently crosses percentile lines during the first 24 months of life as growth becomes less dependent on intrauterine factors and more dependent on genetic factors. During childhood, linear growth has a relatively constant velocity. Most children grow at the following rates: 5.5 to 9 cm/yr from 2 to 4 years; 5 to 8.5 cm/yr from 4 to 6 years; 4 to 6 cm/yr (boys) from 6 years to puberty, and 4.5 to 6.5 cm/yr (girls) from 6 years to puberty. During puberty or adolescence, growth spurts occur (8 to 14 cm per years) due to the synergistic effects of increased secretion of gonadal steroids and GH [26]. In girls, the pubertal growth spurt can occur around age 10 or as early as age 8 for early maturing girls. In boys, the pubertal growth spurt usually appears around the age of 12 years or as early as age 10 in early maturing boys [11].
Definition of short stature
SS is the main reason for referral to pediatric endocrinologists, but the majority of referred children are normal growth variants, display constitutional delay of growth and puberty (CDGP), or have familial short stature (FSS).
SS can be defined as a height 2 SDS below the mean for that age and sex, height less than 2 SDS of the target height (TH), or a significant decrease in height SDS defined as a deflection of at least 0.3 SDS/yr [27-29].
Serial growth monitoring is therefore needed and growth failure is defined as deflection of the growth curve. Furthermore, children with a height below the third percentile for age, sex, and ethnic group on an appropriate growth curve must be accurately evaluated. World Health Organization growth charts can be used for children up to 2 years old, but local growth charts have to be used in older children [30].
Epidemiology
Multiple components such as genetic, prenatal and postnatal factors, life-style, nutrition, and local environmental elements can affect growth in children. Interestingly, the relative contributions of these factors might differ in different populations [33]. The prevalence of pathological cases of SS among children has been reported to range from 1.3% to 19.8% depending on the criteria for referral [34]. In a South Indian District cross-sectional study of school children, the overall prevalence of SS was 2.86% [35]. In addition, the commonest causes of SS in Indian children attending a community hospital were protein energy malnutrition and chronic diseases (53.5%); less frequent causes were normal variants of SS (24.4%), endocrine problems (4.7%), and miscellaneous (5.8%) [36]. A study conducted at a tertiary care hospital in Kolkata reported that 164 of 2,170 children (8%) had SS due to hypothyroidism (29%), chromosomal abnormalities (20%), CDGP (18%), or growth hormone deficiency (GHD) (15%) [37]. Chen et al. [38] reported a SS rate in Shanghai among 6- to 18-year-old children and adolescents of 3.26%. This rate was significantly lower than the percentages reported in Chile in 2005 [39] and in the Wanzhou district of Chongqing in 2014 [40]. In addition, Wang et al. [41] demonstrated that the average detection rate of SS in primary and middle school students was 3.16% in Anhui province. Interestingly, a growth study conducted in Utah demonstrated that among 555 of 79,495 children (0.7%) who had SS, FSS and CDGP accounted for 75% of cases, 14% had underlying organic disease, while only 5% had an endocrine cause. The incidence of GHD (defined as peak GH response to stimulation <10 ng/dL) was 1 in 3,500; 6 girls had Turner syndrome [42]. In a United Kingdom study, Voss et al. [43] reported 180 of 1,400 children (1.3%) with SS at school entry, the majority of whom had FSS, while 32 had organic disease. In Iranian children with SS, FSS accounted for 33.6% of cases, CGDP for 19.7%, idiopathic SS for 11.5%, primary hypothyroidism for 2.5%, and intrauterine growth retardation for 9.8% [44].
Thus, worldwide studies clearly show that FSS, CGDP, and GHD are the most frequent causes of SS. The variation in prevalence of SS described in the different studies is mostly due to differences in the level and nature of health care facilities where the studies were performed, the criteria used to define SS, and the profiles of the study subjects.
Growth monitoring and criteria for referral from primary care
The practice of height measurement to recognize SS in children varies widely across countries [45]. Systematic growth monitoring is not consistently performed throughout the developed world. A mandatory screening program for growth with annual anthropometric evaluation during childhood is performed by primary care physicians in South Korea [46]. Thus, exact trends in height can be determined. Growth monitoring programs also exist in Mexico where careful evaluation of nutrition is a priority and BMI is periodically calculated. In Finland, an accurate height screening program has existed for over 50 years, and a minimum of 20 measurements are performed from the first post-birth evaluation to the age of 12 years [47]. The aim of these screening programs is to identify treatable disorders before symptoms appear. Recorded anthropometric data are then compared with pre-established cutoff standards that characterize normal growth in the same population. An accurate screening program should have sufficient specificity to avoid the referral of healthy children and sufficient sensitivity to identify disorders of growth [27].
In 1998, a Consensus Meeting recognized more appropriate auxological referral criteria based on 3 key screening parameters: the distance between height and TH SDSs, height SDS, and "height deflection" over time [48]. However, these guidelines led to an excess of referral of normal children [49].
Grote et al. [27] published evidence-based guidelines for screening parameters with the highest sensitivities and specificities. In 0- to 3-year-old infants of normal birth-weight, height SDS <–3, or height SDS <–2 on 2 or more cases within 1 year offered the best sensitivity for positive diagnosis, with a low false-positive rate. For 3- to 10-year-olds, the association between the shortness for TH parameter (i.e., height SDS – TH SDS <–2) and height SDS <–2.5 together with the 'height deflection' parameter identified 85.7% of children with Turner syndrome and 76.5% of children with other growth disorders. Analysis of these criteria resulted in development of an algorithm for referral of children with growth disorders, which has recently been updated [50].
Differential diagnosis of SS
Many conditions can occur in association with growth deceleration or SS. In some of these, SS may be the only clinical manifestation. However, the majority of children with SS are healthy, and only some of them have an underlying pathological condition of concern. Prompt identification and management of these children can prevent excessive SS in adulthood.
There are many approaches to classify different causes of SS; causes of SS may be primary, secondary, or idiopathic (Table 1) [51]. Primary growth disorders include defects in the growth plate and several syndromic conditions; the number of these conditions has been expanded in the past few years to include mutations in genes that are able to cause SS without other clinical manifestations. Recent progress in molecular technologies has significantly improved our knowledge of the genetic causes of SS and partially changed how these conditions are approached. The number of known genetic abnormalities that cause growth disorders has increased notably in the last decade, but further research is needed (Fig. 2). Secondary growth disorders are a heterogeneous group of chronic conditions that interfere with the environment of the growth plate and include disorders of the GH-IGF-1 axis and IGF-1 deficiency or resistance, other endocrine and metabolic disorders, chronic systemic diseases, malnutrition, psychosocial disorders, and iatrogenic conditions. Idiopathic short stature (ISS) has no identifiable causes, but children with this diagnosis are characterized by height more than 2 SDS below the mean height for age, sex, and population without any genetic, endocrine, systemic, or nutritional alterations, and includes the diagnoses of FSS and CDGP [52]. In the past few years, numerous conditions previously classified as ISS have been identified as genetic conditions [53].
Evaluation of children with SS
1. Clinical evaluation
The goal of evaluation of children with SS is to identify those who have pathologic underlying causes or hormonal abnormalities. Thorough consideration of several conditions underlying SS and the underlying complex mechanisms is extremely important, making it critical to collect a detailed medical history and perform an accurate physical evaluation of children for an effective diagnostic workup. Initially, there are several fundamental questions to ask as part of the initial evaluation [2]. In particular, information about family history (e.g., consanguinity), pregnancy, and use of assisted reproductive technologies, as well as the anthropometric parameters of children at birth such as their weight, length, and head circumference should be recorded. In addition, gestation and delivery conditions with possible complications should be accurately described. It is also important to focus on the time when growth failure was first observed, and to describe any possible signs and symptoms associated with SS that could reflect a chronic systemic condition, hypothyroidism, precocious puberty, malformations, or neurologic alterations (Table 2).
Several studies have reported that short children are not always evaluated adequately, because there is often a tendency to consider children with SS born to short parents to be normal (FSS), but recent studies have shown that these children and their parents may be short because of an underlying common genetic condition that requires a careful workup.
Thus, an accurate physical evaluation should be performed, including precise auxological measurements. For infants and children up to 24 months of age, length should be measured lying down; for children 2 years and older, height should be measured standing up. The results should be plotted on growth charts that are appropriate for age, sex, ethnic group, and country. Weight should also be measured and BMI calculated to obtain additional information about nutritional status [2]. In fact, feeding is one of the key determinants of growth during infancy and nutritional condition should be a central constituent of any growth appraisal. On the contrary, GH becomes a major determinant of height velocity with an increase in age. Children with isolated GH deficiency have a weight and height that is similar to the normal population at birth, but by the time the child is 2 years old, they will be short (in some cases short length may be evident during infancy) [25].
Assessment of height velocity is an essential component of the evaluation of a child with SS; it requires serial measurements for at least 6–12 months and helps to determine whether the child's height velocity is within the normal range and whether it progressively deviates.
Pubertal stage must be accurately evaluated in all children, and special attention should be paid to evaluating possible dysmorphic features and body disproportions. Body disproportions are evaluated by measuring sitting height and arm span; this ratio is believed to be more reliable and reproducible than upper/lower segment ratios. Other features such as Madelung deformity or mesomelia are indicative of SS caused by mutations in genes encoding short stature homeobox (SHOXM) [54,55], while macrocephaly and lumbar hyperlordosis are suggestive of constitutive activation of fibroblast growth factor receptor-3 (FGFR3) [56].
Another important step is to determine the height range expected for the child's biologic family, which can be obtained by calculating the "target height" based on the heights of both parents, and adjusting this value for sex.
2. Investigations
To carefully assess growth in children with SS, other investigations must be performed on the basis of medical history, physical assessment, and growth evaluation.
In particular, if SS is associated with dysmorphic features, a syndromic condition should be suspected and a genetic test should be performed (Table 2). Karyotyping should be performed in girls with SS even if the typical stigmata of Turner syndrome are not present [57].
In addition, the diagnostic workup of a short child must include radiographs of the hands and wrists to determine bone age, despite this investigation it is not totally standardized. In fact, automated methods for bone age are not available in all countries and are not reliable in children less than 2 years of age. The methods most commonly used are the Greulich and Pyle Atlas [58] and the Tanner-Whitehouse 3 method [59]. Bone age determination is important in a diagnostic workup for SS because it plays a crucial role in defining if the poor growth is associated with delayed or accelerated bone age compared with chronologic age. Delayed or advanced bone age is defined as a bone age that is 2 SDS or more below or above the mean, respectively. Delayed bone age is seen in children with CDGP and in many types of pathologic growth failure, systemic disease, nutritional deficiency, and GHD [52]. On the contrary, advanced bone age is seen in children with precocious puberty and hyperthyroidism. Interestingly, obese children typically have advanced bone age, thus evaluation of radiographs of the hands of these children is not reliable. Advanced bone age, especially in prepubertal children, may also suggest a mutation in the aggrecan (ACAN) gene [60] or identify patients with craniopharyngioma or hypothalamic hyperphagia [61]. Normal bone age is seen in various pediatric conditions, especially in FSS and in younger girls with Turner syndrome. In addition, X-ray of the hands and wrists can help to identify skeletal dysplasia by allowing evaluation of the presence of lucency, triangularization, and pyramidalization of the bones of the wrist, which could suggest SHOX deficiency [54,55], mutation of FGFR3, or mutation of natriuretic peptide receptor 2 (NPR2) (if Madelung deformity has not been recorded). The presence of brachydactyly should raise the suspicion of Indian hedgehog gene (IHH) mutation [62].
Furthermore, apparently healthy children with SS should be investigated for a variety of systemic diseases, especially if they present with delayed bone age. In particular, celiac disease, hypothyroidism, and other inflammatory diseases that result in malnutrition should be excluded.
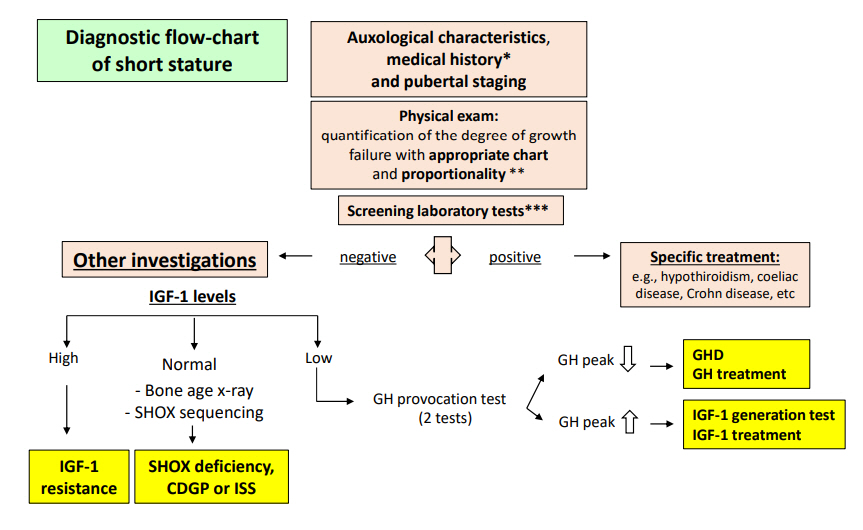
To complete a diagnostic workup for children with SS, some simple blood exams are essential; they should include blood count, C-reactive protein, and erythrocyte sedimentation rate which are useful for identification of inflammatory diseases. Liver and kidney function should be evaluated to exclude renal failure, tubular acidosis, and other chronic systemic conditions [52]. Serum levels of IGF-1 and IGFBP-3 are more important for evaluation of the GH/IGF-1 axis in older children and in children younger than 3 years. Patients with low serum concentrations of IGF-1 for age and sex should be investigated for GHD with stimulation tests. Children with SS and elevated IGF-1 serum concentrations must be evaluated for mutations in the IGF-1 receptor or in the pappalysin 2 (PAPPA-2) gene. PAPPA-2 encodes a metalloproteinase responsible for cleavage of IGFBP-3 and IGFBP-5, a required step in the production of free IGF-1 [63]. Thus, defects in the GH/IGF-1 axis should be suspected and investigated in children with SS, reduced height velocity, and low IGF-1 level [64,65].
Additional tests such as stimulus tests can also be performed in suspected cases of GHD. Two pathologic tests are needed to diagnose GHD. These tests measure the response of the hypothalamus and pituitary gland to various stimuli. The most commonly used stimuli are insulin (insulin tolerance test or ITT), arginine, glucagon, clonidine, L-dopa, and GH-releasing peptide 2. Although these tests are considered gold standards, they have limitations such as poor reproducibility and use of different methods among different laboratories; in addition, there are no universal agreements as to what the normal cutoffs for the different stimuli should be [66-69]. During the past decades, the cutoffs for definition of GH deficiency have been reduced to minimize false-positive tests; institution of these new cutoffs has involved many countries and pediatric endocrine societies. Priming in adolescents is still controversial; some researchers have suggested that all children should have sex steroid priming before stimulation tests to decrease false-positive test results [70].
GHD therefore remains a clinical diagnosis that includes auxological (height SDS and growth velocity), anatomical, radiological (delayed bone age, with some exceptions and pituitary imaging), and laboratory data (IGF-1/IGFBP-3 and GH stimulation tests) in association with clinical expertise.
Cranial magnetic resonance imaging (MRI) to study the pituitary and hypothalamic area should be performed in all children with a diagnosis of GHD after confirmation with GH stimulation tests. If neurologic symptoms or indicative signs of a mass lesion in the pituitary and hypothalamic area are present, cranial MRI must be performed as soon as possible. If diagnosis of GHD is excluded, cranial MRI is not useful. MRI should always be performed in neonates and infants when midline defects are suspected [71].
In the last decade, the development of genetic techniques has led to an increase in the diagnosis of patients with unknown conditions. There are no clear recommendations for how and when to perform a genetic investigation in children with isolated SS. Currently, after the exclusion of nutritional, hormonal, inflammatory or systemic disorders, or skeletal dysplasias and syndromes, genetic causes should be suspected and genetic evaluation performed.
Recent studies have demonstrated that a significant number of healthy short children have a monogenic cause determining their SS, especially those with a diagnosis of FSS. Genes associated with isolated SS include heterozygous variants in genes involved in growth plate development such as SHOX, ACAN, NPR2, NPPC, and IHH [60-62]. These conditions are associated with a variable degree of SS without specific features. Therefore, it is important to use a multiple gene testing approach with next generation sequencing or whole exome sequencing in children with isolated SS without other associated conditions (Table 3). Genes involved in GH production, the GH receptor, or downstream signaling molecules (STAT5B), the effector IGF1 and its receptor, or loss of function mutations in PAPPA-2 that decrease biologically active levels of IGF-1 should also be investigated if indicated. In the future, genetic testing could become a standard step in evaluation of children with isolated and idiopathic SS [63].
A very recent study showed that a polygenic risk score may predict familial SS in Chinese children; researchers identified 10 novel single nucleotide polymorphisms (SNPs) that may help to predict the risk of FSS in Han Chinese children. For the study, the team genetically profiled 1,163 Taiwanese children of Han Chinese ancestry diagnosed with FSS, defined as a height below the 3rd percentile in children who also had one or both parents with a height below the 3rd percentile, a bone age appropriate for chronological age, normal annual growth rate and onset of puberty, and normal biochemistry findings. They identified 10 novel SNPs associated with FSS risk, including SNPs in the known genes COL6A5, LOC105374144, UGT2B17, IQCM, and PGM5P2, with odds ratios (ORs) for FSS ranging from 1.90 to 15.32. A further 9 SNPs previously linked to human height in genome-wide association studies were also associated with FSS, namely DNM3, ANAPC13, LCORL, GPR126, QSOX2, ATF7-ATPG-2, CDK10, CABLES1, and UQCC1, with ORs for FSS ranging from 1.22 to 4.55 [72]. This report paves the way for further analysis using a systems biology approach to improve our knowledge of the determinants of FSS, ISS, and CDGP.
Treatment
The treatment of SS depends on identification of the underlying cause of the disease. GH therapy is recommended as a growth-promoting treatment for many different disorders. In particular, it is widely used in children with GHD because it replaces GH deficiency and is able to increase growth velocity and improve final height and metabolism. The development of recombinant GH (rhGH) resulted in the rapid extension of clinical trials to evaluate the effects of GH in various conditions characterized by non-GH deficient SS. The most frequent condition treated with GH is GHD; other growth-related indications for GH treatment are Turner syndrome, SHOX gene deficiency, Noonan syndrome, Prader-Willi syndrome, growth failure associated with chronic renal insufficiency, SS in children born small for gestational age who do not show catchup growth, and ISS (Table 4) [73].
Treatment with a pharmacological dose of GH, initially indicated for GHD, has been proposed for various non-GHD conditions. This treatment is based on empiric observation of growth acceleration after GH administration. However, GH therapy in Down syndrome, neurofibromatosis, 18q deletion syndrome, gastrointestinal disorders as Crohn disease, bone dysplasia (hypochondroplasia, achondroplasia, hypophosphatemia, and rickets), pulmonary defects such as cystic fibrosis, hematologic problems and endocrine abnormalities such as precocious puberty, and congenital adrenal hyperplasia is not approved by the U.S. Food and Drug Administration (FDA) or the European Medicines Agency [73-75].
Recent advances
A standardized approach to identify children who will respond to treatment is lacking, but genomic, proteomic, and metabolomic techniques could help in the future [72].
One recent development is "Macimorelin," an oral ghrelin agonist, that is a new stimulus for GH production that was approved by the FDA in 2017 for diagnosis of GHD in adult patients. Indeed, a multicenter, open-label, randomized, twoway crossover trial was designed to validate the efficacy and safety of a single oral dose of Macimorelin for diagnosis of GHD in adult patients compared with the ITT. In this study, it was demonstrated that oral Macimorelin was easy to administer, well tolerated because it does not depend on hypoglycemia, and only required the collection of 4 venous blood samples after administration. Finally, the results obtained were reproducible and it was safe to perform compared to ITT [76,77]. High reproducibility (94%) and elevated estimated sensitivity (92%) and specificity (96%) using a GH cutoff of 5.1 ng/mL were obtained relative to ITT [76,78]. Therefore, the oral Macimorelin test may improve the diagnostic accuracy of stimulation tests for GHD and be better tolerated, but there are no published data using this stimulus test in children.
Recombinant GH is currently approved for daily use in GHD and has been shown to improve final height and metabolism with few adverse effects. Daily injection, however, can be painful, demanding, and/or stressful. Therefore, new formulations such as long-acting GH with a longer duration of action have been developed to improve adherence to GH treatment, as fewer injections may increase acceptance, tolerability, and therapeutic flexibility, and can potentially improve treatment outcomes. Long-term surveillance registries and trials to assess the efficacy and safety of longer-acting formulations in children and adults have been planned [79,80].
C-natriuretic peptide (CNP) is another important regulator of longitudinal bone growth that acts through natriuretic peptide receptor B (NPR2). CNP signaling via NPR2 in chondrocytes inhibits the mitogen-activated protein kinase signaling pathway by down-regulating FGFR3 signaling. Gain-of-function mutations in the FGFR3 gene result in achondroplasia and hypochondroplasia. Several authors have reported that CNP analogues may be useful in these conditions81 [82], and a clinical trial in children is in progress.
Further studies are necessary to address various issues associated with the diagnosis and treatment of children with SS, for example international standardization of the GH and IGF-1 assays, standardization of GH stimulation tests, and determination of different cutoffs for diagnosis of GHD for various pubertal stages. In addition, new biomarkers derived from metabolomic approaches for diagnosis of GHD and evaluation of the efficacy of rhGH treatment are needed to improve diagnosis and early treatment.
Conclusions
Growth is a complex process in which nutrition, hormones, genetic factors, and environmental factors play important roles. For decades, the GH-IGF-I axis has been a central focus, but not all underlying mechanisms have been elucidated. Recent findings in molecular and cellular biology as well as clinical genetics have highlighted a wide group of other mechanistical systems that regulate skeletal growth and an accompanying vast array of genetic disorders that can cause defects in normal growth.
The application of genetic techniques has improved our understanding of the molecular basis of SS and has resulted in the discovery of new causes of SS and the underlying cellular mechanisms. Detailed characterization of the phenotype of a pediatric or adolescent patient is necessary to putatively identify a specific condition and to define the workup (hormonal, image and genetic) that should be performed to decipher the cause of the SS.
 , stimulatory;
, stimulatory;  , inhibitory.
, inhibitory.