Introduction
Rickets is a disease of growing bone that is due to unmineralized matrix at the growth plates1). Vitamin D deficiency is the most common cause of rickets2,3), but any condition that disturbs bone mineralization, including distal renal tubular acidosis (RTA), can cause rickets4,5). Distal RTA is commonly accompanied by hypercalciuria and nephrocalcinosis, resulting in calcium deficiency6).
Distal RTA is a disease of defective urinary acidification, which is caused by insufficient net acid excretion by the kidneys7). The disease is characterized by alkaline urine in the presence of systemic metabolic acidosis. In its severe form, affected patients are very acidotic and volume-depleted, with marked hypokalemia but otherwise normal renal excretory function. Levels of calcium and phosphate are usually normal, but growth is poor, and rickets is common in untreated cases because of leaching of minerals from the bones in an attempt to buffer the acidosis. Bone resorption by osteoclasts is increased in acidosis8).
We describe a case of distal type RTA with renal calcification in children during follow-up of a rickets patient who presented with clinical manifestations of short stature, failure to thrive, recurrent vomiting, dehydration, and irritability.
Case report
A 6-year-old boy was hospitalized because of delayed speech, failure to thrive, and weakness in both legs. He was born at 32+0 weeks' gestation by cesarean section and weighed 1.8 kg. He was slow to gain weight and had constipation since he was 6 months old. At 4 years of age, he could not walk very well, and both legs were thin on presentation. He had two elder sisters and one elder brother, and no one had experienced similar problems.
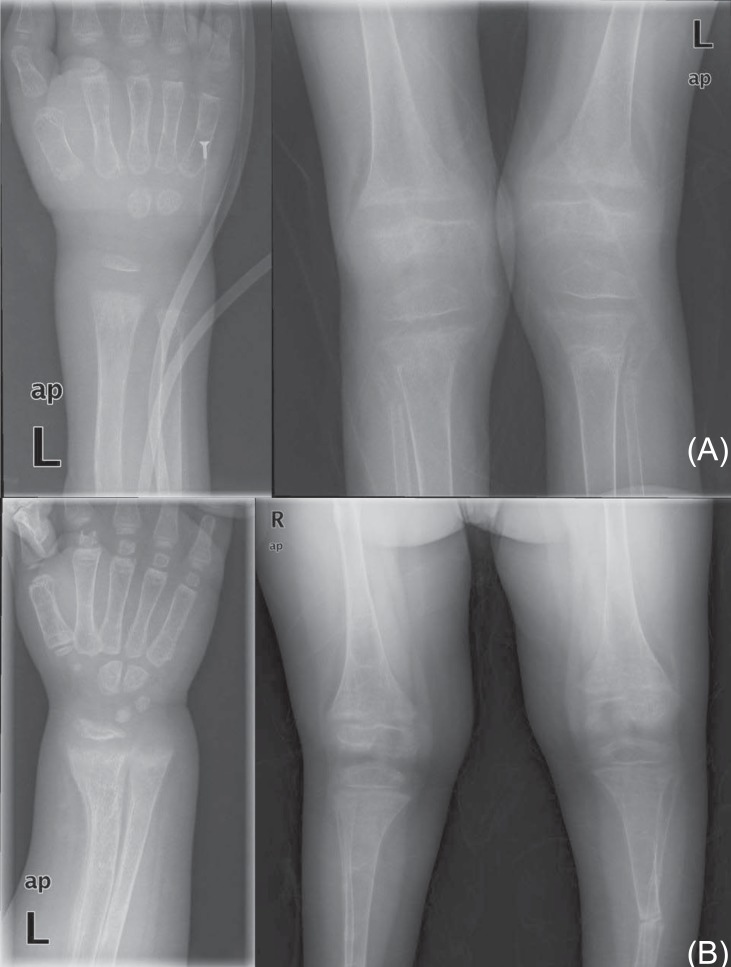
His vital signs were unremarkable. He was short in stature, measuring 95 cm (<3rd percentile) and weighing 15.6 kg (<3rd percentile). A complete blood cell count was normal. Electrolyte battery showed hypokalemia and hyperchloremia. Serum calcium was within the normal range, serum phosphate was decreased, and alkaline phosphatase was markedly increased. Arterial blood gas analysis showed mild metabolic acidosis, with a normal anion gap (Table 1). Urine electrolytes showed decreased chloride excretion, with a positive urine anion gap and marked hypercalciuria in 24-hour urine. Thyroid function tests were normal. Levels of 25-hydroxy (25-OH) vitamin D and 1,25-dihydroxy (1,25-OH) vitamin D were decreased. Parathyroid hormone levels were within the normal range. A skeletal survey showed diffuse osteopenia, fraying in the distal radius and fracture of the left tibia and both fibulae (Fig. 1). Lumbar bone mineral density was decreased in dual energy X-ray absorptiometry (DEXA). A brain magnetic resonance imaging was normal. Brainstem auditory evoked potential showed no significant sensorineural hearing loss, with a threshold of 20 dB bilaterally. Electromyography and nerve conducting velocity showed no unequivocal electrophysiological abnormalities.
He was managed with oral calcium, vitamin D, and spironolactone, with intravenous potassium chloride replacement. After a few days, his hypokalemia and metabolic acidosis were corrected, and he was discharged.
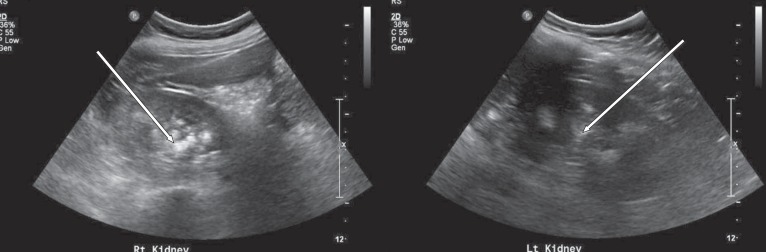
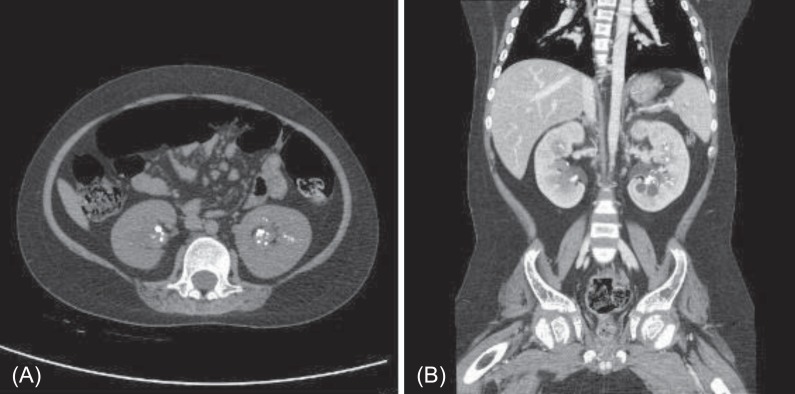
At 11 years of age, he was hospitalized again because of vomiting, irritability, and poor oral intake. Despite long-term calcium, potassium, and vitamin D therapy, he was still short in stature. He measured 110 cm (<3rd percentile) and weighed 32 kg (25th to 50th percentile). Electrolyte battery showed hypokalemia and hyperchloremia. Chemical batteries showed hypocalcemia and hypophosphatemia, and alkaline phosphatase was still increased. Arterial blood gas analysis showed metabolic acidosis with a normal anion gap (Table 1). Urinalysis showed alkaline urine indicative of metabolic acidosis, with a positive urine anion gap. A 24-hour urine analysis showed improved but persistent hypercalciuria. Levels of 25-OH vitamin D and 1,25-OH vitamin D were within the normal range. A skeletal survey showed persistent diffuse osteopenia, fraying in both the distal radius and the ulna and fracture of the left tibia and fibula (Fig. 1). The lumbar bone mineral density was still decreased in DEXA but increased compared with the previous study. Kidney ultrasonography showed increased echogenecity in renal parenchyma bilaterally (Fig. 2), but a 99mTc-dimercaptosuccinic acid renal scan was normal, and there was no evidence of decreased excretory function. Abdomen and pelvis computerized tomography showed multiple calcified nodules in both renal parenchymae and thinning of the renal cortex (Fig. 3).
He was managed for distal RTA with fluid therapy and a large amount of bicarbonate (5-10 mEq/kg/day) and potassium citrate (5 mEq/kg/day). Dehydration and acidosis were completely corrected. He started to gain weight two weeks after the commencement of the therapy, and his electrolyte levels returned to normal. Hypercalciuria was improved, but the calcium/creatinine ratio was still high. The patient is now 12 years old. He is still of short stature and shows rachitic changes.
Discussion
RTA is a clinical syndrome that causes hyperchloremic metabolic acidosis due to a disorder of urine acidification. The acidification of urine in the distal tubule primarily depends on acid-base exchange transporters in intercalated cells. There are three related processes: provision of H+ and HCO3- ions from H2O and CO2 by cytosolic carbonic anhydrase II (CA II), excretion of H+ ions into the collecting tubule by vacuolar H+-ATPase, and excretion of HCO3- ions into the blood by the HCO3-/Cl- anion exchanger (AE1)9). Impaired function of any of these components can cause a defect in urine acidifica-tion, and the severity of functional defect depends on the importance of affected component10). Genetic mutations causing functionional impairment of the components were recently discovered, with studies reporting that mutations in the gene encoding cytosolic CA II are associated with mixed-type distal and proximal RTA and with cerebral calcification11,12). Mutations in the gene that encodes AE1 usually result in distal RTA13). Mutations in the ATP6V0A4 and ATP6V1B1 genes encoding the a4 and B1 subunits of vacuolar H+-ATPase can cause inherited distal RTA and sensorineural hearing loss14-16). Most patients with distal RTA have hypokalemia because the inability to excrete H+ is compensated for by increased K+ secretion.
In this case, the patient had chronic hyperchloremic metabolic acidosis, a normal anion gap, alkaline urine with a positive urine anion gap, hypokalemia, and nephrocalcinosis. Although these findings are consistent with distal RTA, there was no evidence of sensorineural hearing loss or cerebral calcification. A mutation of AE1 was suspected, but we did not perform a genetic study to identify the mutations accompanying the RTA. The current case also appears to be incomplete distal RTA17) because the metabolic acidosis was not very severe. An NH4Cl loading test would be required to confirm the diagnosis, but we did not perform it. This could be a limitation of our study.
The aim of distal RTA treatment in children is not only correcting biochemical abnormalities, but also improving growth and preventing kidney stones and skeletal abnormalities. The basis of the treatment is alkali replacement. Potassium citrate or sodium citrate is preferable to bicarbonate because citrate salt can correct hypocitraturia and prevent nephrolithiasis. Potassium supplementation and K+-sparing diuretics, such as spironolactone or amiloride, are necessary to manage the hypokalemia. If the distal RTA is accompanied by rickets, calcium, and vitamin D replacement are required. In this case, despite treatment, the patient's vertical growth and bone mineral density remained poor. His short stature may be due to another cause or due to insufficiency of the treatment. Some reports have described delayed bone age and decreased growth velocity in distal RTA18). Phosphate supplementation could improve clinical features in vitamin D-resistant rickets with RTA19). So, in cases that fail to respond to calcium and vitamin D replacement therapy, further tests should be conducted to determine the source of the short stature. And if needed, treatment with growth hormones and bisphosphonate should be considered20).