Introduction
Adrenal hypoplasia congenita (AHC) is a developmental disorder of the human adrenal gland which results in significant hormonal deficiencies and is lethal if untreated. Deletions or mutations of the gene DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), that is now designated NR0B1 (nuclear receptor subfamily 0, group B, member 1), cause the X-linked form of AHC1). The DAX-1 gene, located on Xp21, consists of 2 exons. It can also occur as a part of contiguous gene deletion syndrome together with glycerol kinase (GK) deficiency and/or Duchenne muscular dystrophy (DMD)2). It is classically known to cause hypogonadotropic hypogonadism as a puberty disorder3). However, few cases have reported its association with central precocious puberty even though its mechanism has not been identified4,5).
Here we report a male patient with X-linked AHC and contiguous gene deletion of DAX-1, GK and IL1RAPL1 who developed central precocious puberty instead of hypogonadotropic hypogonadism.
Case report
The 9 year and 4 months old boy who has been diagnosed as having primary adrenal insufficiency was noticed to have enlarged testes at the age of 45 months. He was born at 41+3 weeks' gestation, weighing 3.4 kg by spontaneous vaginal delivery. He was presented with poor weight gain and dark skin pigmentation on whole body at the age of 2 months. On admission, his initial sodium level was 118 mEq/L, potassium level was 5.6 mEq/L and urine sodium was 92 mEq/L, suggesting mineralocorticoid deficiency. The urinary level of 17-ketosteroid collected for 24 hours was low as 0.47 mg/day (reference, less than 0.5 mg/day). Other initial results before treatment were as followed: 17-hydroxyprogesterone (17-OHP), 20.5 ng/mL (reference, less than 3 ng/mL); cortisol, 43.4 µg/dL (reference, 2.8-23 µg/dL); adrenocorticotropic hormone (ACTH), 250 pg/mL (reference, 10-60 pg/mL); renin, 0.63 ng/mL/hr (reference, 2.35-37 ng/mL/hr) (Table 1). Under the impression of congenital adrenal insufficiency, he started taking hydrocortisone and florinef. During follow-up, he had several episodes of admission due to adrenal crisis associated with febrile illnesses.
When he became 8 months of age, he could only roll over. He couldn't sit alone and had difficulty in creeping. We performed Bayley developmental test three times (at the age of 13 months, 25 months, and 40 months) and the results showed persistent severe mental retardation.
When he was 45 months, his height was 110.9 cm (z score, 2.17) and his weight was 19.2 kg (z score, 1.40). His testes were 4 mL in volume bilaterally with Tanner stage 2 pubic hair. Bone age acceleration was also observed (chronologic age 45 months, bone age 66 months). Since gonadotropin-releasing hormone (GnRH) stimulation test showed the luteinizing hormone (LH) peak of 8.26 IU/L (Table 2), we started GnRH agonist treatment under the diagnosis of central precocious puberty.
At 55 months of age, we performed ACTH stimulation test because of his atypical manifestations such as mental retardation and the initial relatively high cortisol level which are not usually found in congenital adrenal hyperplasia. In ACTH stimulation test, the peak cortisol level at 60 minutes was 7.40 µg/dL, suggesting cortisol deficiency. The level of 17-OHP was also lower than normal and the ratio of 17-OHP/cortisol at 60 minutes was less than 0.1 which was not compatible with congenital adrenal hyperplasia due to 21-hydroxylase deficiency (Table 3).
Genetic study showed DAX-1 deletion, confirming X-linked AHC. Deletion of the genes of GK and IL1RAPL1 was also observed, which we call 'Xp21 contiguous gene deletion syndrome'. Deletion of IL1RAPL1 gene is known to cause X-linked intellectual disability. His younger brother who was born one year after him, was also diagnosed as Xp21 contiguous gene deletion syndrome with same mutation.
It is unusual to have central precocious puberty in the case with DAX-1 deficiency. Subcutaneous injection of GnRH agonist (leuprolide acetate) started at 46 months. Follow-up of GnRH stimulation test at 50 months showed suppressed LH responses. After 1 year of GnRH agonist therapy, we stopped GnRH agonist injection for 6 months and reevaluated the pubertal status. On repeated GnRH stimulation test, the peak LH level showed pubertal level, therefore we restarted the GnRH agonist therapy.
For evaluating GK status, we checked urine glycerol and it was slightly increased (Cr, 10.58 mmol/mol; reference, not detected). Hypertriglyceridemia of 370 mg/dL (reference, 31-108 mg/dL) was also consistent finding to GK deficiency (Table 1).
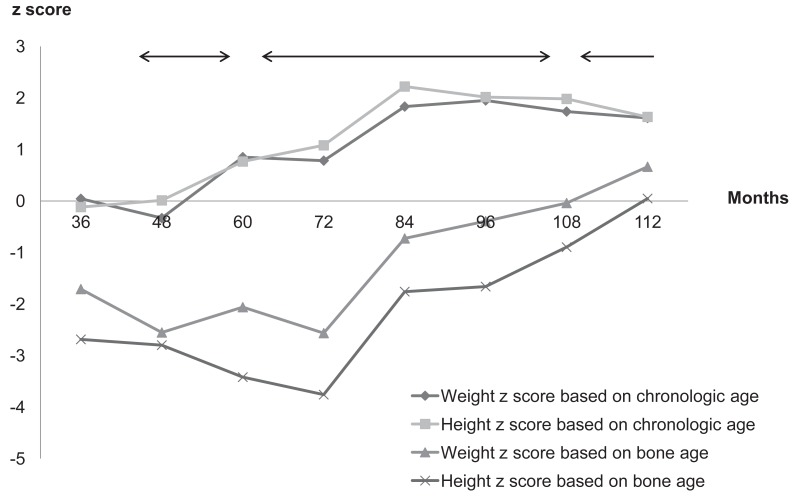
Now he is 9 year and 4 months old (bone age, 11 year and 2 months) with height of 143.8 cm (z score, 1.63), weighing 45 kg (z score, 1.61) (Fig. 1). The improvement in growth z score based on bone age reflects that he is a good responder in GnRH agonist therapy as well as hydrocortisone (20.5 mg/m2/day) and florinef (0.1 mg/day) medication.
Discussion
X-linked AHC is one of the most common causes of primary adrenal failure. It was initially described by Sikl6) in 1948. In 1994, mutation in the nuclear receptor gene DAX-1 was reported as the causative genetic abnormality. Patients with X-linked AHC frequently develop severe salt-wasting with glucocorticoid and mineralocorticoid insufficiency in infancy and it is generally related to hypogonadotropic hypogonadisim3,7). This is because the adrenal glands lack the permanent adult cortical zone. The remaining cells are termed "cytomegalic" because they are larger than typical fetal adrenal cells and contain characteristic nuclear inclusions from cytoplasmic invaginations8).
It is useful to differentiate 21-hydroxylase deficiency (congenital adrenal hyperplasia) with X-linked AHC because each could have different clinical courses as well as different mode of inheritance.
In our case, initial renin level was decreased which was not compatible with typical finding in untreated X-linked AHC patient. This was because the blood test was done after 2 hours of hydration.
Gene analysis was done because of atypical developmental delay and decreased serum level of 17-hydroxyprogesterone on ACTH stimulation test.
Genetic test revealed Xp21 contiguous gene deletion syndrome at Xp21 locus which involved DAX-1, GK, and IL1RAPL1 genes.
X-linked AHC is usually associated with hypogonadotropic hypogonadism because DAX-1 mutations probably impair gonadotropin production by acting at both the hypothalamic and pituitary levels9). But the onset of puberty could be variable from arrested or absent puberty10,11) to precocious puberty4,5,12). Precocious puberty is generally defined as the appearance of secondary sex characteristics before age 8 years in girls and before 9 years in boys13). Only few cases reported the relationship between X-linked AHC and precocious puberty. Wittenberg14) described four male cousins with X-linked AHC in 1981 and they showed varying degree of virilization. Their responses to GnRH test was prepubertal which suggested gonadotropin-independent precocious puberty (peripheral precocious puberty). Domenice et al.12) reported 2-year-old Brazilian boy with DAX-1 mutation whose first clinical manifestation was isosexual gonadotropin-independent precocious puberty. He presented with pubic hair, enlarged penis and testes and advanced bone age. Steroid replacement therapy induced a decrease in testicular size and testosterone levels to the prepubertal range. It was hypothesized that chronic excessive ACTH levels may stimulate the Leydig cells, leading to gonadotropin-independent precocious puberty in some boys.
Gonadotropin-dependent precocious puberty (central precocious puberty) in X-linked AHC patients was also reported4,5,15). Katsumata et al.15) reported the first association of X-linked AHC with presumed central precocious puberty in 1997. The patient was found to have a novel frameshift mutation caused by a 2-nucleotide (AC) insertion at nucleotide 1007, and duplication of the following 11 nucleotides. At the age of 6 months, he developed pubic hair, and at 1 year and 3 months of age, his mean testicular volume increased to 3.5 mL. He was presumed to have central precocious puberty, based on the increased urinary excretion of gonadotropins and testosterone.
Loke et al.4) reported the first case of central precocious puberty due to complete deletion of DAX-1 gene. At 6 years of age, he was noticed to have testicular volumes of 4 mL bilaterally, a penile length of 8.5 cm and pubic hair of Tanner stage 2. His response to GnRH stimulation was pubertal suggestive of gonadotropin-dependent precocious puberty.
Recently, Durmaz et al.16) reported three boys in one large family with novel nonsense Gln208X mutation in the amino terminal domain of the DAX-1 gene presented different pubertal status. Two boys developed spontaneous puberty that failed to progress at similar ages, whereas the other boy developed precocious puberty at 10 months of age. The exact mechanism of the mutation on puberty was not known.
Therefore, we need to keep monitoring pubertal status during follow up of the patients with X-linked AHC although the mechanism might be multifactorial which will need further studies.